Updated labs on 6 months of therapeutic dosing Intramuscular + Intranasal IM.
Just a reminder, I am taking at least 10X the weekly dose than what most everyone is taking orally. (1 week post dose trough sirolimus level/AUC as reference).
Based on above MK study update, looks like 99% of people are taking 10mg/week or less ORALLY, which would likely produce <1 ng/mL at 1 week trough.
For example, if your 1 week trough Sirolimus is 0.75 ng/mL, then my Jan 25, 23 trough of 7.5 ng/mL is 10X the dose. My 11.1 ng/ml trough Aug 3 would be almost 15X the typical daily oral dose.
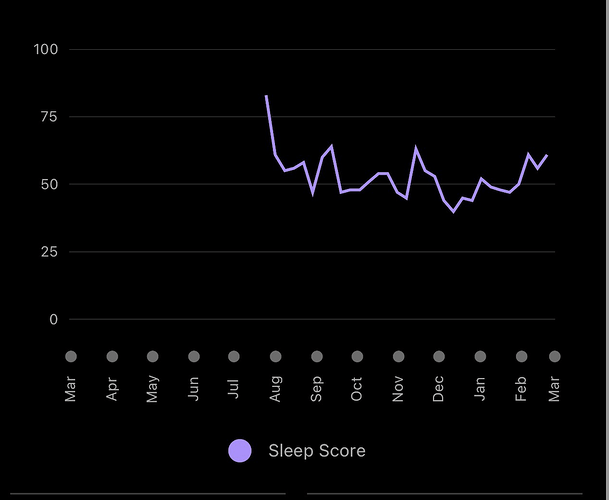
I am currently in washout (procedure, doc wanted all meds stopped, so out of abundance of caution), although feeling normal at 6 months, still able to do daily high exercise, even though full on anemic. However there seems to be a pretty strong negative sleep signal (Garmin watch sleep score). It’s definitely been trending downward on this protocol…more fragmented sleep, I feel more wired at night (take weekly dose in the AM). From summary below, I started high dose rapamycin in July 2022. There is a definite trend downward throughout the 6 month intervention (last dose Jan 18, 23)
At time of writing, my sleep score has been trending back up on washout (per above summary), although I have added a confounder, since started taking high dose slow release melatonin at night, so wrecked the control experiment. I definitely think rapamycin at high doses hampers sleep, which needs a hack, as sleep is HUGE to long term health
Will be doing a baseline of new labs, new gut updated kit microbiome, and then restarting.
MAC INTRAMUSCULAR & INTRANSAL RAPAMYCIN LABS.pdf (55.8 KB)
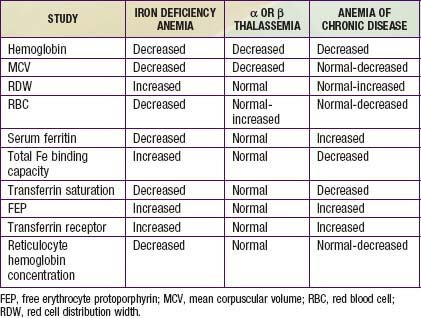
What’s most notable in the summary is how my TG/FG/hbA1c are relative proxies for trough sirolimus level. The glucose markers change not that significant/interesting in this 6 month update. I’ve had no weight change, no overt signs I’m turning diabetic at 6 months, although a small bump in a still extremely low fasting insulin. I’m strict keto/OMAD, so my AUC glucose/insulin is normally extremely low; starting at the extreme opposite of diabetic/insulin resistant phenotype. Low inflammation, low iron, low insulin, superior physical health…certainly a resistant phenotype. What would happen to me several years on this protocol?
Far more interesting are the lipids and understanding ASCVD risk…something of still raging debate. Does perturbation of lipids by rapamycin raise ASCVD risk? But mTOR inhibition (longevity) comes only with high dose therapeutic, so the all- cause-mortality (ergo lifespan) paradox/dichotomy!
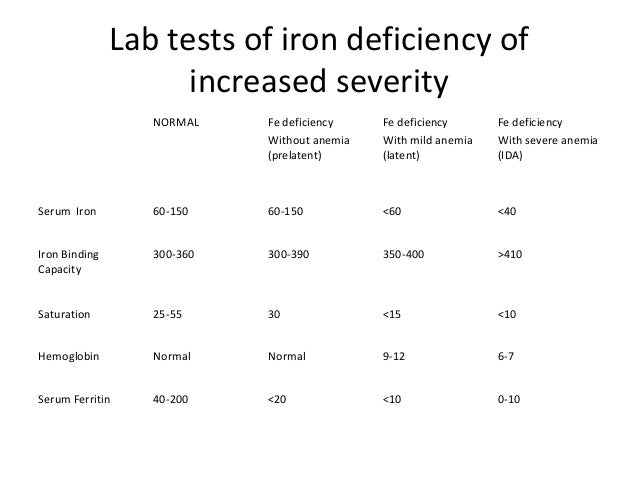
The iron and immuno markers are still at very low normal. Iron saturation is still less 20%, although Ferritin did come up a bit (I stopped donating blood when I started this protocol back in July 2022). The very low iron levels pre therapeutic dose rapamycin put me at much higher “clinical anemia risk” on this protocol, but my long term low iron dumping (and high exercise, oxygen efficiency) seems to have produced a physiological adaptation even to 6 months of high dose rapamycin. Overall, still very much in clinical anemia state, but perhaps reached bottom and uptick. But with washout, will never know trajectory until next and longer term therapeutic intervention. My entire iron journey is suggestive that modern humans are at MUCH HIGHER iron levels than physiologically necessary. For most of evolution and in the 3rd world, people were and are infected with hookworms and parasites that removed iron! Well that’s no longer the case, and modern humans keep building and building iron stores as we age.
“Radioisotope studies with chromium 51-tagged red blood cells have shown that patients with heavy hookworm infection can lose up to 250 ml, or a quarter of a liter of blood, daily, and up to 29 mg of iron in the gastrointestinal tract, thus leading to direct iron-deficiency anemia.”
As iron dumping is one of my central longevity hacks, my n=1 certainly begs the question as to whether modern guidelines should be revisited, separate from the many longevity benefits of low iron.
Diving right in, how is that my TC/LDL/apoaB are relatively UNCHANGED FROM PRE RAPAMCYIN PROTOCOL?? Is this n=1 OR something unique about method of delivery, namely bypassing of 1st pass metabolism by way of IM + IN?
A seminal paper on the impact of therapeutic ORAL dosing rapamycin (10mg/day in general, although individually tailored, trough Sirolimus > 5ng/mL) on renal transplant patients is quite interesting and serves as relative protocol comparison of oral dosing. There are NO healthy persons cohorts taking therapeutic dose rapamycin…I am n=1.
Effects of sirolimus on plasma lipids, lipoprotein levels, and fatty acid metabolism in renal transplant patients
https://www.jlr.org/action/showPdf?pii=S0022-2275(20)30049-3
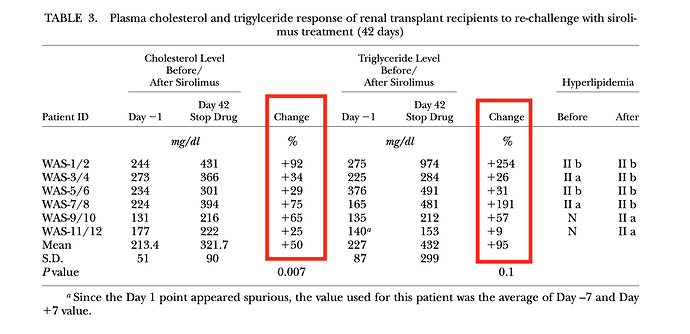
They did a first 14 day dosing discovery trial, and then a second phase of the study, they gave patients daily dosing for 42 days, then measured various lipid markers.
In the 42 day trial, average change in TC and TG was increased 50% and 95%, respectively, although quite a bit of inter-person variability. “In general, triglyceride level were highly responsive to sirolimus dosage. Throughout the entire 6-week treatment period (42 days), sirolimus had no effect on HDL-C levels”
Compare and contrast with my lipids…TC was essentially unchanged, whereas TG did rise by approximately 100%, and HDL remained essentially unchanged vs pre rapamycin dosing baseline.
What about LDL-C? Although the paper didn’t discuss, I extracted the data in a summary table.
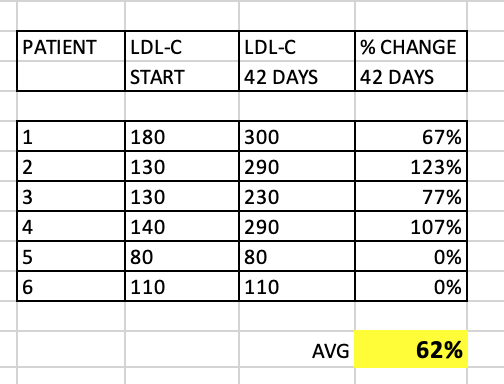
As you can see, quite a significant overall increase in LDL-C (62% average), although some patients had very little change at 42 days. These patients with little change in LDL-C still had significant change in TC/TG. Why would my TC, and for that matter, my LDL-C, remain relatively UNCHANGED?
What about apoB? The study measured apoB-100, which is the same as apoB.
As we know, apoB and LDL-P have a much stronger association to ASCVD, than LDL-C. I extracted the data from the study figures, and prepared a summary table:
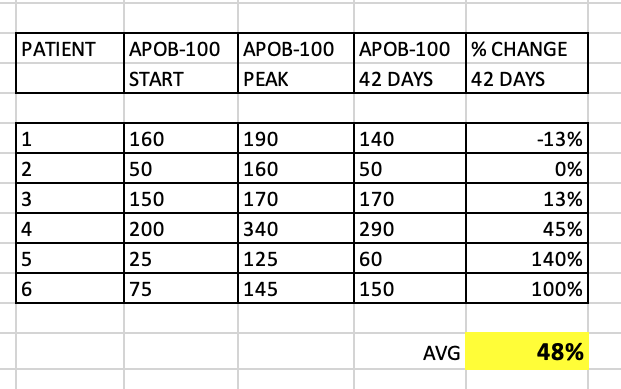
Several patients had a peak in apoB, but the 42 day apoB wildly varied between patients. Some patients had only a small increase, and some over 100%. What’s very interesting is that even the patients with no overall change in LDL-C at 42 days…still had > 100% increase in apoB! Quite the discordance between these 2 lipid markers in this small cohort.
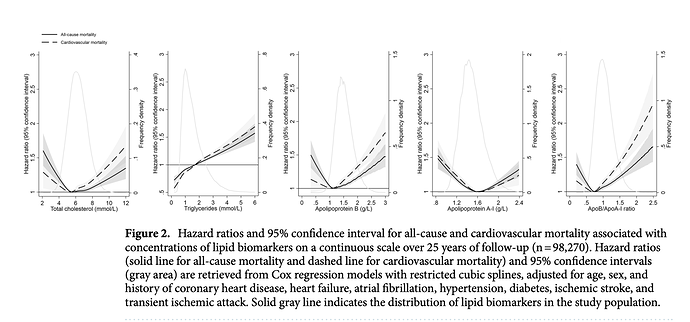
I have 3 apoB measurements during my 6 months, and my level remained essentially unchanged, as did my apoB/apoA1 ratio, itself a marker used in ASCVD. Although a very recent (2021) study showed a U shaped curve to apoB in all cause mortality.
https://www.nature.com/articles/s41598-021-03959-5
Quite interestingly, my apoB and apoB/apoA1 are right at lowest HR. And even with therapeutic high dose rapamycin, my TG of 120mg/dL (1.35 mmol/L) puts me at neutral HR, although the lower the better.
And very interestingly, my LDL-C remained relatively unchanged over the course of the high dose intervention. Why are my TC, LDL-C and apoB resilient to this therapeutic rapamycin dosing…n=1 genetics or method of delivery? Seems less likely n=1 uniqueness given above cohort data that, even though there is clearly VERY potent genetic inter-person variability to rapamycin dosing. Is my liver/genetics that lipids unique to these 6 renal transplant patients? What are the chances I would have a complete outlier of all 3 of TC/LDL-C/apoB unchanged on therapeutic dosing? The protocol? The greater significance long term?
The study also measured apoC-II and apoC-III.
“ApoC-II and apoC-III are important protein components of VLDL and HDL. The plasma levels of apoC-II were typically low and did not change appreciably during the course of the study. In contrast, the initial values of apoC-III were substantial, and increased significantly between day 1 and day 42. Since apoC-II is an activator and apoC-III is an inhibitor of lipoprotein lipase (LPL), these results provide a reasonable explanation for the substantially lower LPL activity in these renal
transplant patients (20–70%) compared with normolipidemic controls.”
“Triglyceride hydrolysis by lipoprotein lipase (LPL), regulated by apolipoproteins C-II (apoC-II) and C-III (apoC-III), is essential for maintaining normal lipid homeostasis. During triglyceride lipolysis, the apoCs are known to be transferred from very low-density lipoprotein (VLDL) to high-density lipoprotein (HDL)”
The study also measured free fatty acids (FFA) as an attempt to further under TG dysregulation and clinical implications. FFA are derived from triacylglycerol by cleavage lipase (in the body).
“These results were supported by measurements of total free fatty acid levels, which indicated considerable expansion of this pool. Taken together, these results support the view that sirolimus enhances the action of hormone sensitive lipase (HSL) and perhaps also inhibits LPL. These effects are the opposite of those mediated by insulin, suggesting that sirolimus may induce hypertriglyceridemia via an insulin-dependent signaling pathway. If the drug interferes with insulin-stimulated triglyceride storage in adipocytes, this could lead to increased release of FFAs into the circulation, their increased uptake by the liver, and increased hepatic secretion of VLDL triglycerides. Alternatively, sirolimus may also decrease FFA oxidation leading to increased FFA availability.”
We know from other animal studies, therapeutic dosing rapamycin massively alters TG metabolism.
https://sci-hub.se/10.1016/j.metabol.2006.01.017
“Rapamycin treatment resulted in more than a 2-fold increase in plasma triglycerides (TG), whereas no differences were observed in plasma cholesterol between RAPA and control groups. Very low density lipoprotein and low-density lipoprotein particles isolated from guinea pigs treated with RAPA were larger in size and contained more TG molecules than particles from control animals. Interestingly, plasma free fatty acids and fasting plasma glucose were 65% and 72% higher in the high-RAPA group than in control. These results suggest that RAPA interferes with TG metabolism by altering the insulin signaling pathway, inducing increased secretion of very low density lipoprotein. In the current study, high plasma TG levels were associated with larger VLDL*** particles carrying an increased amount of TG when secreted by the liver and, subsequently, a larger LDL particle, which was also shown to have higher TG content as compared to the controls. Furthermore, it was observed that the increase in the mean LDL size was mainly due to decrease in smaller, denser fraction of the LDL. This effect of RAPA can be considered positive because smaller LDL is more susceptible to oxidation and increases the risk of coronary heart disease by 3-fold”***
So similar findings to the human renal transplant study, although a fascinating sub analysis of the particle size…MUCH larger on high dose rapamycin. Translatable to human ASVCD risk stratification on high dose rapamycin? Does this trump other lipid markers dysregulated, eg FFA?
And what about elevated FFA…impact on non rapamycin taking cohorts?
“Most obese individuals have elevated plasma levels of free fatty acids (FFA) which are known to cause peripheral (muscle) insulin resistance. They do this by inhibiting insulin-stimulated glucose uptake and glycogen synthesis. FFAs also cause hepatic insulin resistance. They do this by inhibiting insulin-mediated suppression of glycogenolysis. Individuals who are unable to do this (probably for genetic reasons) eventually develop type 2 diabetes. FFAs have recently been shown to activate the IkappaB/NFkappaB pathway which is involved in many inflammatory processes. Thus, elevated plasma levels of FFAs are not only a major cause of insulin resistance in skeletal muscle and liver but may, in addition, play a role in the pathogenesis of coronary artery disease.”
Some references on FFA:
Free fatty acids, cardiovascular disease, and mortality in the Multi-Ethnic Study of Atherosclerosis
Serum free fatty acids are associated with severe coronary artery calcification, especially in diabetes: a retrospective study
Elevation of Free Fatty Acids Induces Inflammation and Impairs Vascular Reactivity in Healthy Subjects
Plasma Free Fatty Acids and Risk of Heart Failure
https://www.ahajournals.org/doi/10.1161/CIRCHEARTFAILURE.113.000521
Plasma Free Fatty Acids and Risk of Heart Failure
https://www.ahajournals.org/doi/pdf/10.1161/circheartfailure.113.000521
Elevated plasma free fatty acids increase cardiovascular risk by inducing plasma biomarkers of endothelial activation, myeloperoxidase and PAI-1 in healthy subjects
Significance of plasma free fatty acid level for assessing and diagnosing acute myocardial infarction
https://sci-hub.st/10.2217/bmm-2019-0291
“Conclusion: An elevated FFA level is an independent risk factor and independent diagnostic marker for AMI.”
In reviewing these papers, the general consensus is that elevated FFA is an independent risk factor for ASCVD. Does the FFA on high dose rapamycin translate to these studies on non rapamycin cohorts? In non rapamcyin cohorts, it certainly suggests higher ASCVD risk.
Lastly, let’s take a closer look at apoB and ASCVD risk. More recently the CVD research community has largely moved on from LDL-C as ASCVD prognosticator, and instead looks at small LDL, oxLDL, and LDL-P (LDL particle number, of which apoB is proxy).
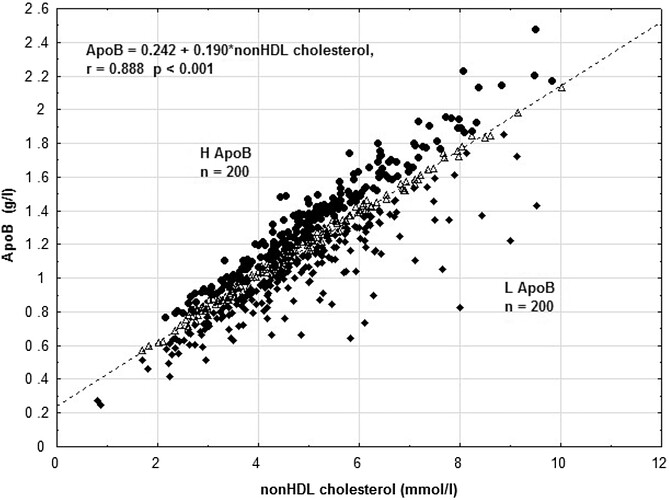
Some reference marker apoB correlation graphs, as most people don’t do a full lipoprotein NMR to get LDL-P…so what can use as good proxies?
https://sci-hub.se/https://doi.org/10.1016/j.jacl.2017.01.020
https://www.dovepress.com/getfile.php?fileID=21661
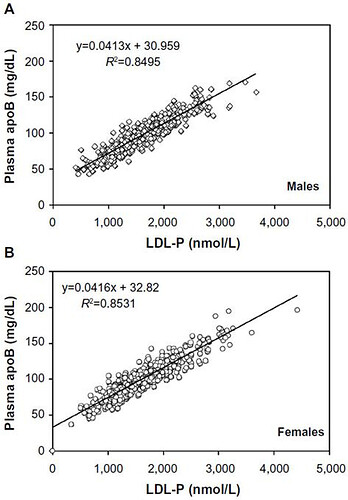
As you can see, apoB is highly correlated to LDL-P.
Attia has many good podcasts on cholesterol and ASCVD that many people follow:

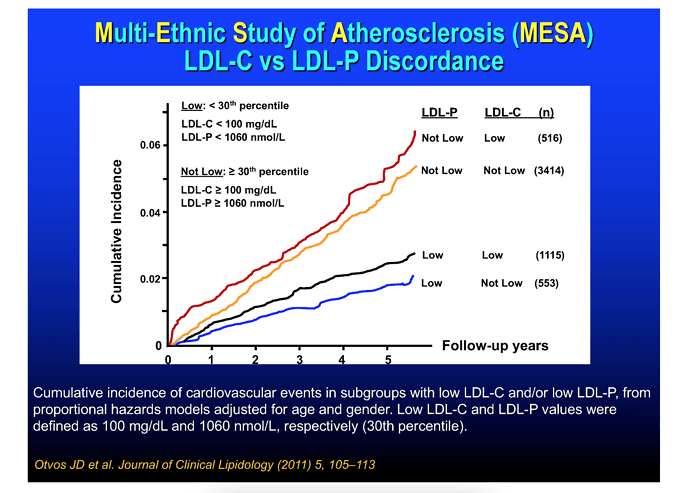
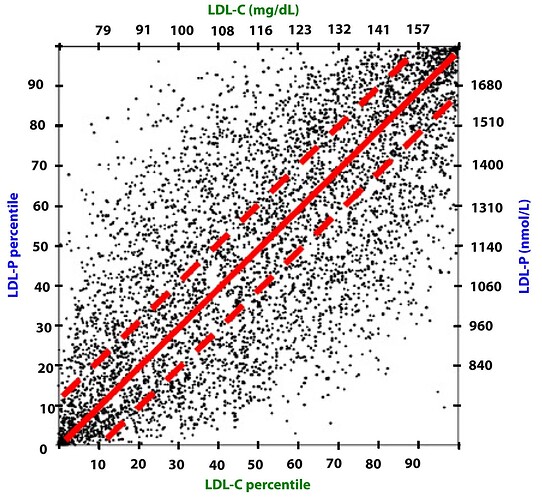
These graphical summaries show clearly a couple of things. Firstly LDL-C and apoB can indeed by DISCORDANT. But LDL-P trumps LDL-C in stratifying risk. We see clearly here that one can have high LDL-C, yet low LDL-P, the net is LOW ASCD risk. In fact, in the 2nd graph, the blue line shows in those with lowest LDL-P, the HIGHER LDL-C is LOWER risk??
Clearly, LDL-C is mostly irrelevant as singular ASVCD risk marker. One must look at the entirety of a person’s phenotype, risk factors, and sub lipoprotein classes TRUMP LDL-C (LDL-P, oxLDL).
In my own n=1 case, I’ve had highish LDL-C even before starting this protocol…looking back from when I started tracking labs 6+ years ago, my LDL-C was around 130, remained similar levels 6 yrs on a PLANT FAT based ketogenic/OMAD diet, and remained stable on 6 months of high dose rapamycin.
I recently (last 2 months) had a cardiologist consult update to baseline ASCVD risk (5 yrs since last update). He rested Lp(a), even though it was very low as from a historical US lab test (one cannot change Lp(a), genetic marker), he wanted his own result (confirmed very low < 10 on upper lab range of 75). He added several ultrasound imaging tests, including femoral, aortic, and peripheral arterial (legs). The results came back CLEAR. Although I’ve had a slight increase my CAC, which is exactly consistent with chronic endurance running (runners paradox), my elevated LDL-C has NOT apparently increased my ASCVD. I also had a full body CT scan as preventative diagnosis, and as an aside, I asked the radiologist to look for whole body arterial calcification. His report “I hope to have this vascular health later in life”. In totality of my clinical makers and phenotype, my cardiologist relayed “whatever you are doing, keep on doing it”. He has not written a statin script, but he’s ITCHING to do it…it’s the standard of care, he needs to cover his butt. I mean, I came to see him and I have elevated LDL, so it’s documented. So clearly, I am n=1 that elevated LDL-C does NOT necessarily correlate with elevated ASVCD and the literature does show there exist higher LDL-C, lower ASCVD risk phenotypes.
Before I restart rapamycin, I will however, be doing FFA , oxLDL, and full NMR lipoprotein labs to baseline these markers in advance of restarting (also as a check of my baseline LDL-P, and how it correlates to apoB. One cannot assume 100% that apoB is LDL-P, sdLDL, or oxLDL proxy; must have the data to check these values (clearly we know genetics/phenotype has huge impact n=1). I can then track these deeper insight markers on therapeutic dosing, and if I do it long enough, re-evaluate ASCVD status with further imaging.
From Attia:
-
LDL-P (or apoB) is the best predictor of adverse cardiac events, which has been documented repeatedly in every major cardiovascular risk study.
-
LDL-C is only a good predictor of adverse cardiac events when it is concordant with LDL-P; otherwise it is a poor predictor of risk.
-
There is no way of determining which individual patient may have discordant LDL-C and LDL-P without measuring both markers.
So my LDL-C and apoB remain UNCHANGED on high dose rapamycin? And what if the raised TG produced larger particle size? Does it mean I didn’t raise my ASCVD risk?
In this washout period, I am experimenting as curiosity, the use of a natural supplement to see how it impacts my LDL-C (perhaps apoB, since I have baseline markers)
Effect of bergamot on lipid profile in humans: A systematic review
“Based on data, 75% of studies showed a significant decrease in total cholesterol, triglycerides and LDLc. The decrease in total cholesterol varied from 12.3% to 31.3%, from 7.6% to 40.8% in LDLc and from 11.5% to 39.5% in triglycerides. Eight trials reported HDLc increase after intervention with bergamot.”
If there is a significant drop in my lipids (will also check apoB), I may continue with this supplement longer term, and then baseline my labs as “pre rapamycin”. Of course, will discuss with my cardiologist to make a final decision.
In summary, my 6 months of therapeutic dose rapamycin has produced a lipids phenotype which is apparently clinically different than therapeutic ORAL dosing rapamycin. Is this superior and clinically significant as rapamycin longevity intervention? Elevated TG definitely remains CENTRAL to both modes of delivery…again, clinically significant? I need more data, longer duration to tease out. UNCHARTED WATERS
The renal transplant/therapeutic rapamycin/lipids study definitely confirms very clearly: everyone will have a UNIQUE response to rapamaycin dosing. And as I’ve stated several times in the past, if you’re not measuring your FULL lab markers such as trough Sirolimus (the morning just before your NEXT DOSE that same morning), lipids, glucose, A1c, full CBC, iron panel, immuno markers, etc, you have NO idea the impact of your dosing protocol. (In fact, for the renal transplant study, they actually had to tweak daily dosing to maintain therapeutic levels, whilst minimizing massive lipid dysregulation in some patients: genetics). Of course, things get amplified in therapeutic dosing protocols re lipid dysregulation, but there are very strong n=1 GENETICS at play.
For those taking CYPA34 inhibitors to boost rapamycin, I understand the rationale to cheaply boost AUC, but as controlled experiment, I doubt very much you can 100% reproduce the AUC spike at every dose/GFJ bolus (variation between fruit, dose volume, speed of gastric remodelling based on say prior meal contents, impact of supplements taking, time of day, etc etc). Certainly, IF you are regularly testing trough Sirolimus, then all is good re getting reproducibility of dosing data insight. As you can see from my data, I also cannot get exact repeated trough Sirolimus at supposed “equal dosing”, although my goal is simply to get above therapeutic dosing (> 5 ng/mL); always achieved.