jnorm
#402
I think the biggest problem with the MMC results for now is the huge question marks surrounding translation. The physiology of roundworms is so different from us that any sort of superficial analysis of how these interventions affect them will fall short. Even getting down to the level of cellular assays, due to the much greater cell type diversity in humans, it’s almost impossible to predict how a result might translate.
At least with mice we can show that an intervention modulated inflammation, fibrosis, markers of active immunity (roundworms don’t even have an active immunes system), etc, and due to the conservation of these processes, along with the various organs/tissues, stem cell niches, endocrine regulation, etc, it’s much more likely that results in mice will translate to humans, at least compared to roundworms
I think one potential path forward is to standardize one or a few epigenetic assays for C. elegans. They don’t generally regulate their DNA by tagging cytosine bases with methyl groups, so DNA methylation assays are not appropriate. But if we obtained their histone modification data instead, we could study how the principal components evolve with age/in response to the drugs and how these changes relate to the patterns seen in the methylation PCA data of mice and humans (similar to what Fedichev is doing).
2 Likes
Acetylation (my favourite PTM - Post Translational Modification) is quite dynamic so I think it would be really hard to monitor it might need some form of flourescence or something to monitor it as it is going on. Obviouly it can be tested for, but at that point the cell is probably broken in some way so what you see is not going to be exactly what you would get in vivo.
My approach with acetylation is to try macroscopic interventions and see what macroscopic changes occur.
In the mean time I think C. Elegans has relatively little value because of things like not having senescent cells and the sensitivity to the health of E. Coli (food). On the other hand I am willing to fund the experiments being proposed with Indolepropionamide where we have three people willing to put up USD100 towards the USD400 costs because Indolepropionamide is such an interesting molecule with so little data and a little data would be good value even it is simply that the worms don’t have a lifespan change.
2 Likes
jnorm
#404
Generally you’re measuring bulk levels, at which point the real-time changes become less important.
From a mechanistic perspective, however, the real time changes are very interesting.
Looking, for example, at the differentiation of stem cells. We start with a cell which has complex 1 inhibited and depends upon pyruvate to maintain redox status. Then (and there is some link here to coming out of a hypoxic environment) it moves towards differentiation, NF kappa B is turned right up creating a substantial citrate flux from the mitochondria and large scale acetylation occurs to enable differentiation. It is at this point that differentiation can fail and the cell enter senescence. It would be nice to understand this at a lower level, but I don’t think it can be measured properly.
Acetyl-CoA is also part of the cell division process and I would like to fully understand the process whereby cells move from a single stem cell to a daughter cell and continuing stem cell.
chatGPT has given me a bit of an answer on this.
Acetyl-CoA is more than a metabolic intermediate; during proliferation it acts as a three-way hub that couples carbon availability to the nuclear, cytosolic and membrane-building events that let a cell duplicate itself.
| Pillar |
What happens |
Why acetyl-CoA is indispensable |
| 1. Epigenetic ignition of the cell-cycle engine |
The first wave of genes needed for G1/S entry—including the yeast G1 cyclin CLN3 and hundreds of “growth” genes—are switched on only when their promoters become hyper-acetylated. That burst of acetyl marks is limited by how much acetyl-CoA is present in the nucleus; boosting acetyl-CoA drives histone acetylation and cell-cycle entry, while starving it keeps chromatin closed and the cycle off. |
• Acetyl-CoA directly fuels GCN5/CBP/p300 histone acetyl-transferases.
• Local nuclear production by ATP-citrate lyase (ACLY) or acetyl-CoA synthetase (ACSS2) ensures that histone acetylation can spike even when cytosolic levels are low. (PubMed , Nature) |
| 2. Building blocks for two daughters |
Every round of division roughly doubles plasma-membrane area, ER and Golgi surface, and the lipid sleeve that envelopes segregating chromosomes. All of that membrane is built from fatty acids and sterols that start as cytosolic acetyl-CoA. |
• Cytosolic acetyl-CoA (mainly from ACLY) is converted by ACC → FASN → elongases/desaturases into phospholipids and neutral lipids.
• Knocking down ACLY, blocking ACC, or inhibiting FASN stalls cells in G1 or kills them because membrane mass cannot keep pace; a 2024 study even uncovered a “lipid checkpoint” in early G1 that arrests the cycle when fatty-acid synthesis (and thus acetyl-CoA consumption) is inadequate. (PubMed , Nature) |
| 3. Acetylation as a molecular timer |
Many cell-cycle regulators themselves are acetylated. For example, Cyclin A must be acetylated at specific lysines before the APC/C can ubiquitinate and destroy it, ensuring orderly mitotic exit. Tubulin and several CDKs are similarly controlled. Because acetyl-CoA is the acetyl donor, its concentration influences how quickly these timers tick. |
• When acetyl-CoA is limiting, cyclin acetylation and degradation slow, lengthening mitosis; excess acetyl-CoA has the opposite effect. (PubMed ) |
How the three arms are coordinated
-
Nutrient sensing → acetyl-CoA synthesis. Growth-factor signalling (PI3K–mTOR) up-regulates ACLY and ACLY’s citrate supply; AMPK and PGC-1α fine-tune ACSS2 and mitochondrial export, ensuring that carbon flows to acetyl-CoA only when energy is adequate.
-
Spatial pools matter. Nuclear ACLY/ACSS2 complexes generate an “on-site” pool for chromatin, while cytosolic ACLY feeds ACC/FASN for lipids. Compartmentalisation lets a cell prioritise gene activation even when membrane synthesis must pause, and vice-versa.
-
Feedback to checkpoints.
- Histone acetylation of cyclin-D/E promoters activates CDK4/6 → Rb phosphorylation → S-phase entry.
- The G1 lipid checkpoint senses incomplete fatty-acid synthesis (a proxy for low acetyl-CoA flux) and activates PERK–ATF4 → p21, blocking CDK activity until lipid supply recovers.
Big-picture view
-
Signal Acetyl-CoA levels rise in response to carbon and growth signals.
-
Set chromatin Local nuclear acetyl-CoA spikes open proliferation genes.
-
Supply biomass Cytosolic acetyl-CoA is channelled into fatty-acid/cholesterol synthesis so the cell can physically double.
-
Synchronise timing Acetyl-CoA-dependent acetylation of cyclins/CDKs/microtubules ensures each phase ends only when the previous one is metabolically “paid for.”
Because all three roles depend on the same metabolite, perturbing acetyl-CoA synthesis or utilisation (e.g., with ACLY inhibitors) simultaneously starves chromatin remodelling, lipid supply and cell-cycle timing—explaining why such enzymes are promising anti-proliferative drug targets in cancer and why acetate supplementation can sometimes rescue dividing cells under metabolic stress.
In short: acetyl-CoA acts as the metabolic throttle of cell division, translating carbon abundance into the chromatin state, macromolecular building blocks and molecular timers that collectively allow one cell to become two.
We have 4 people willing to put the money into this. Just waiting for a response from Ora on this one to get it started.
I thought I would ask chatGPT for a bit more on the process of differentiation of stem cells. I suppose it is not really relevant to this thread as C Elegans adult cells don’t divide.
Hence I have put that here:
2 Likes
Looks like donations through vida útil.io have been removed. Anyone know why or when they’re coming back?
Sponsor an intervention (Ora Biomedical) | Lifespan.io
2 Likes
adssx
#409
This is a question for @Mitchell_Ora 
2 Likes
Hello!
To my knowledge, nothing has been changed regarding our fiscal partnership with Lifespan.IO. I will pass along to Ben to have a look.
Thanks for the heads up!
4 Likes
adssx
#413
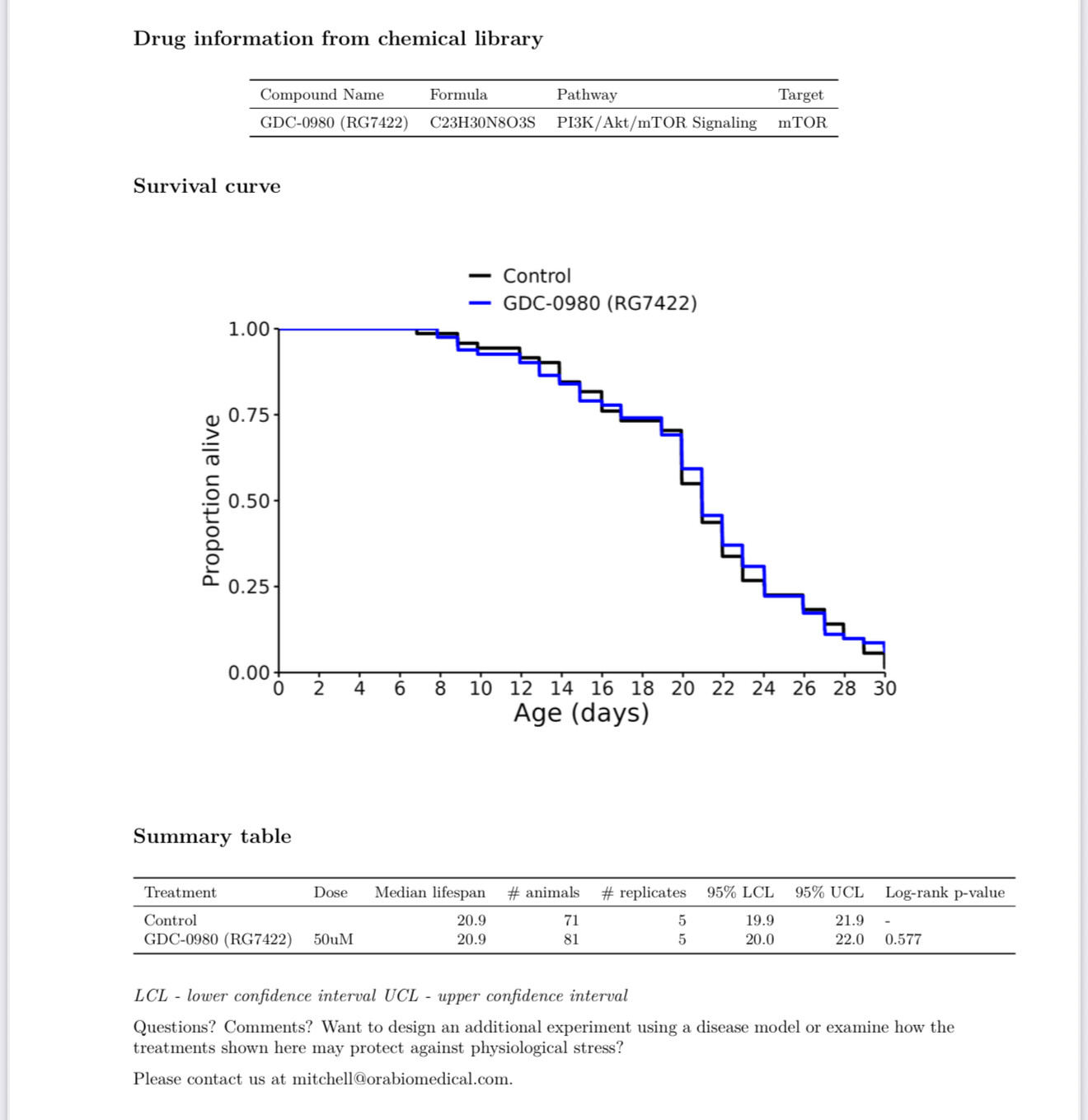
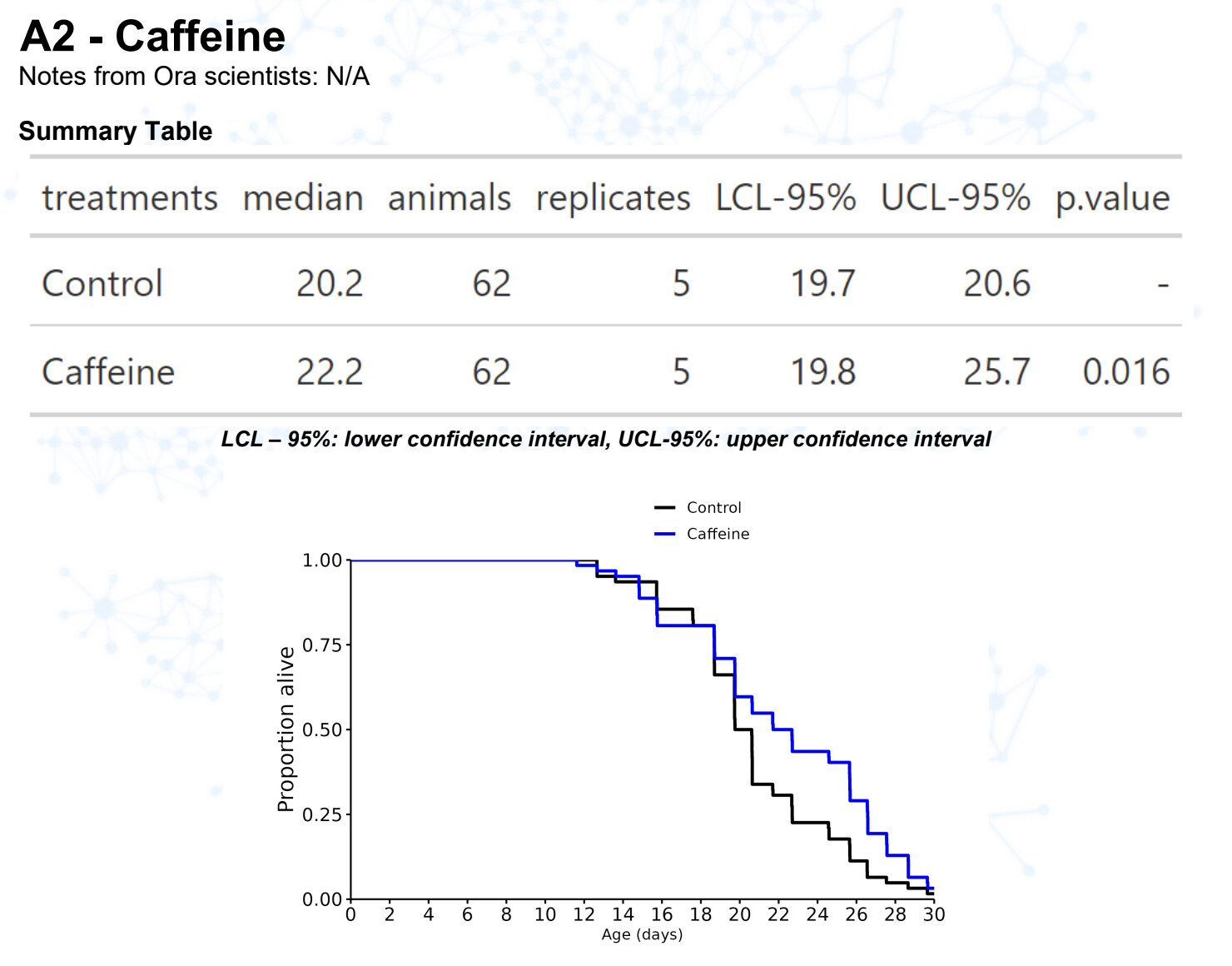
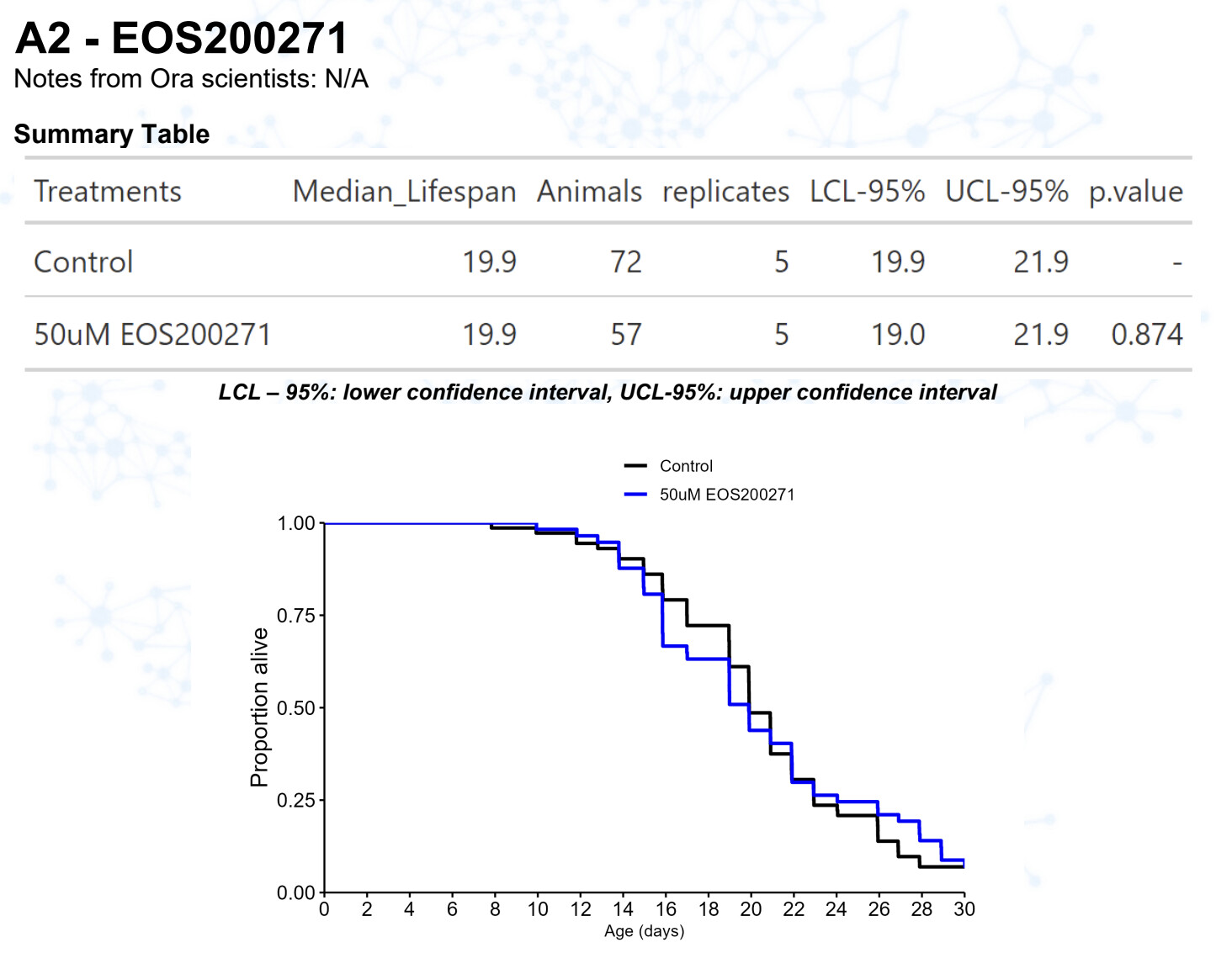
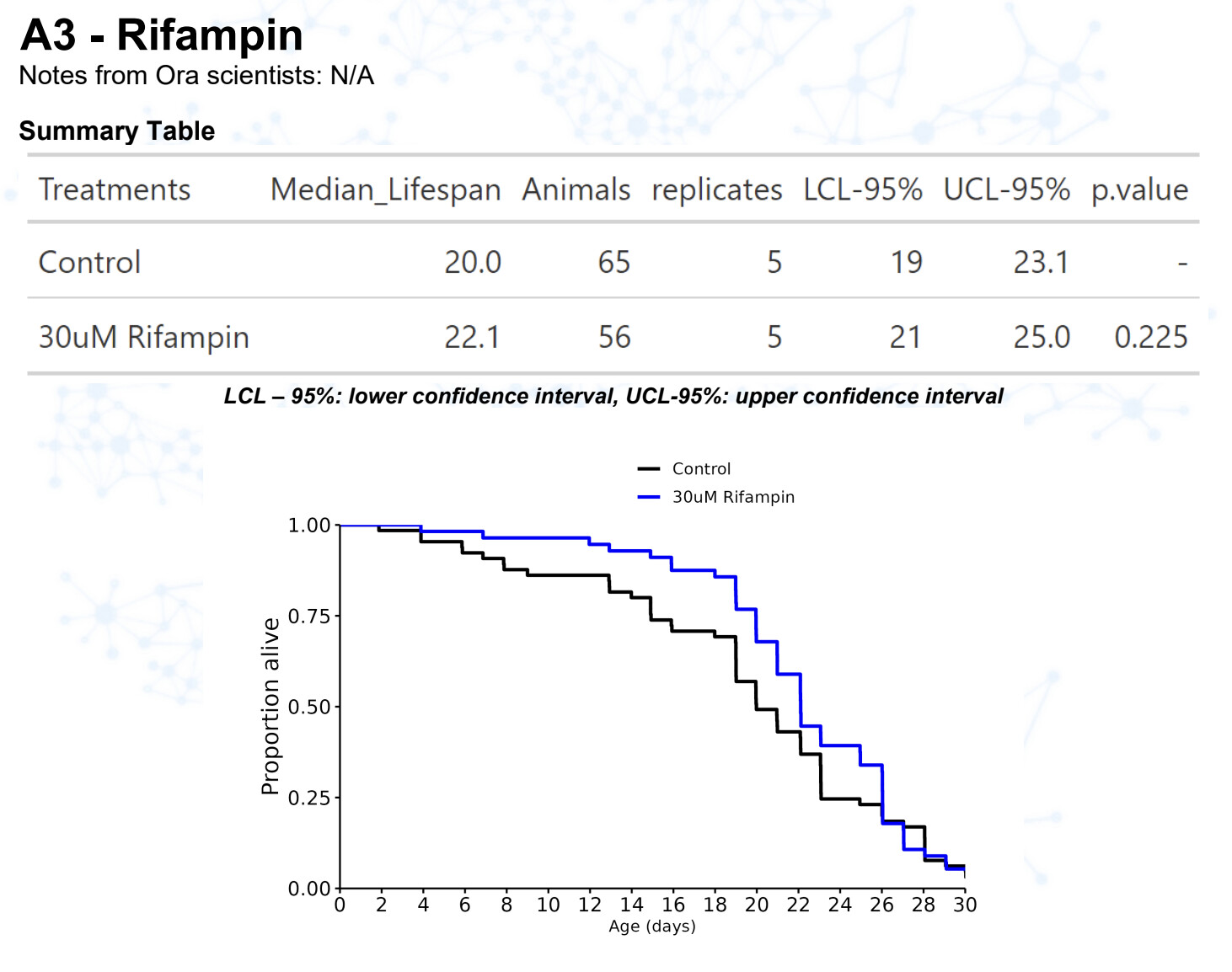
New results:
Caffeine
EOS200271 / PF-06840003 (from this Alzheimer’s paper: https://www.science.org/doi/10.1126/science.abm6131 )
Rifampicin / rifampin
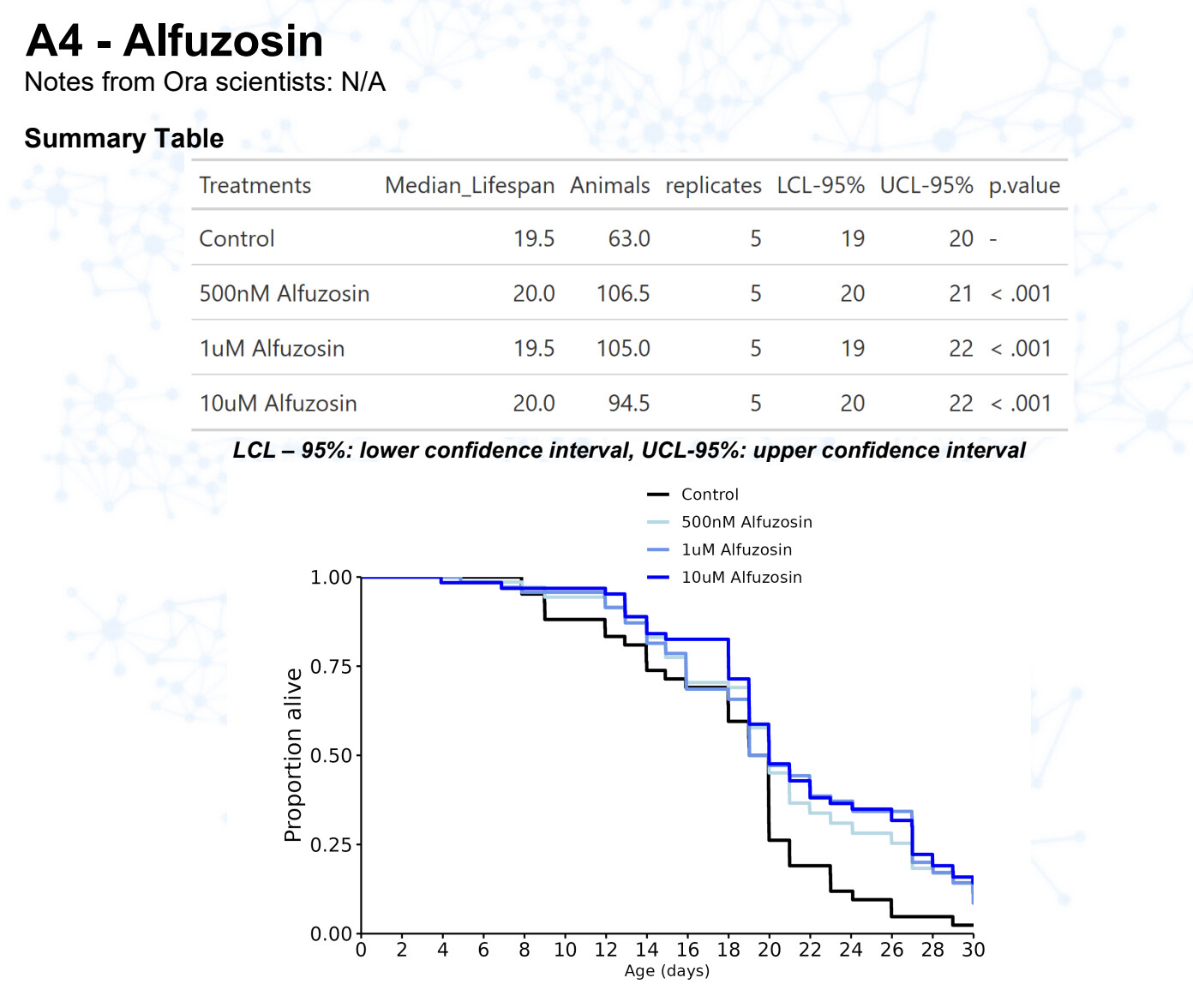
Alfuzosin
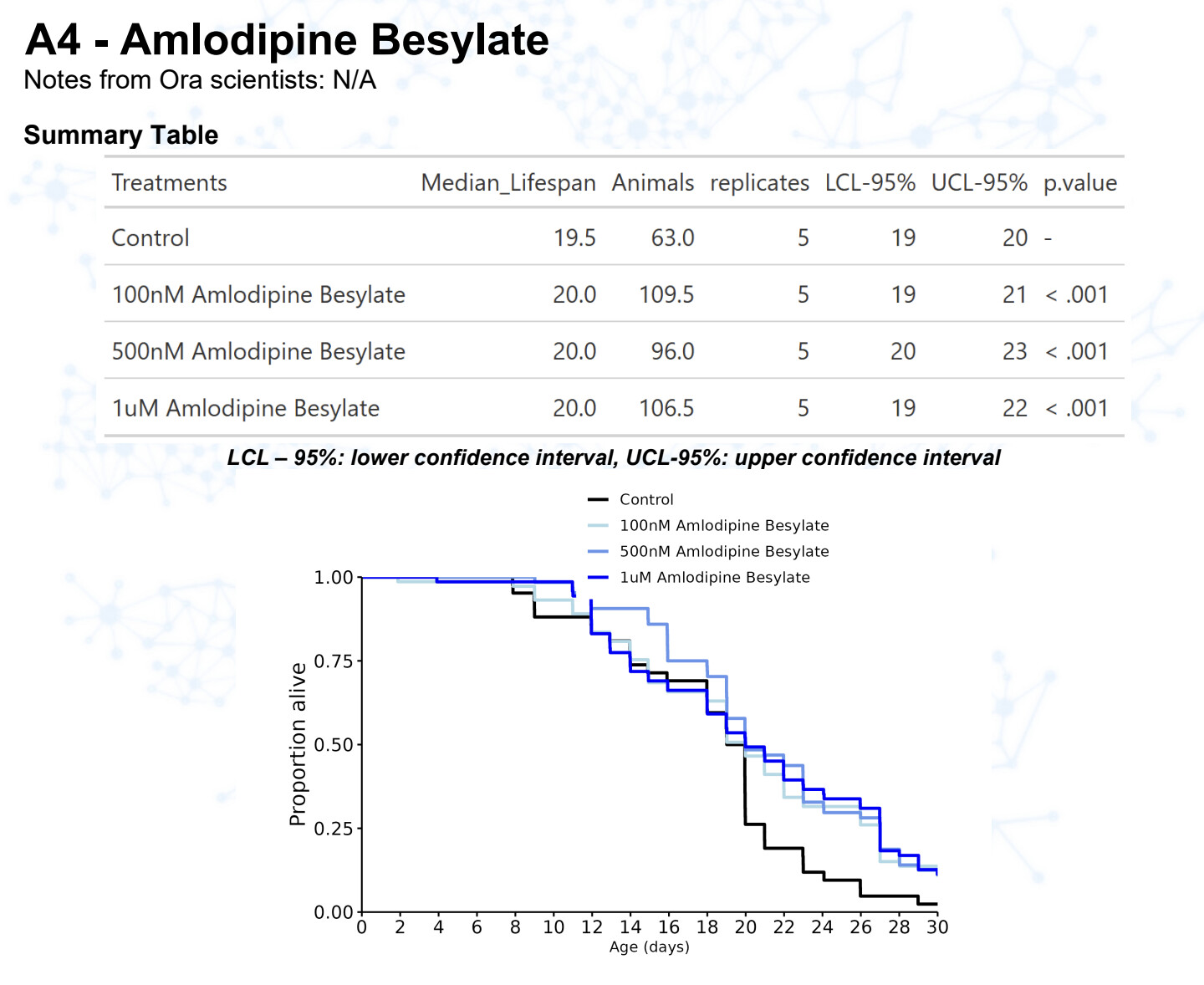
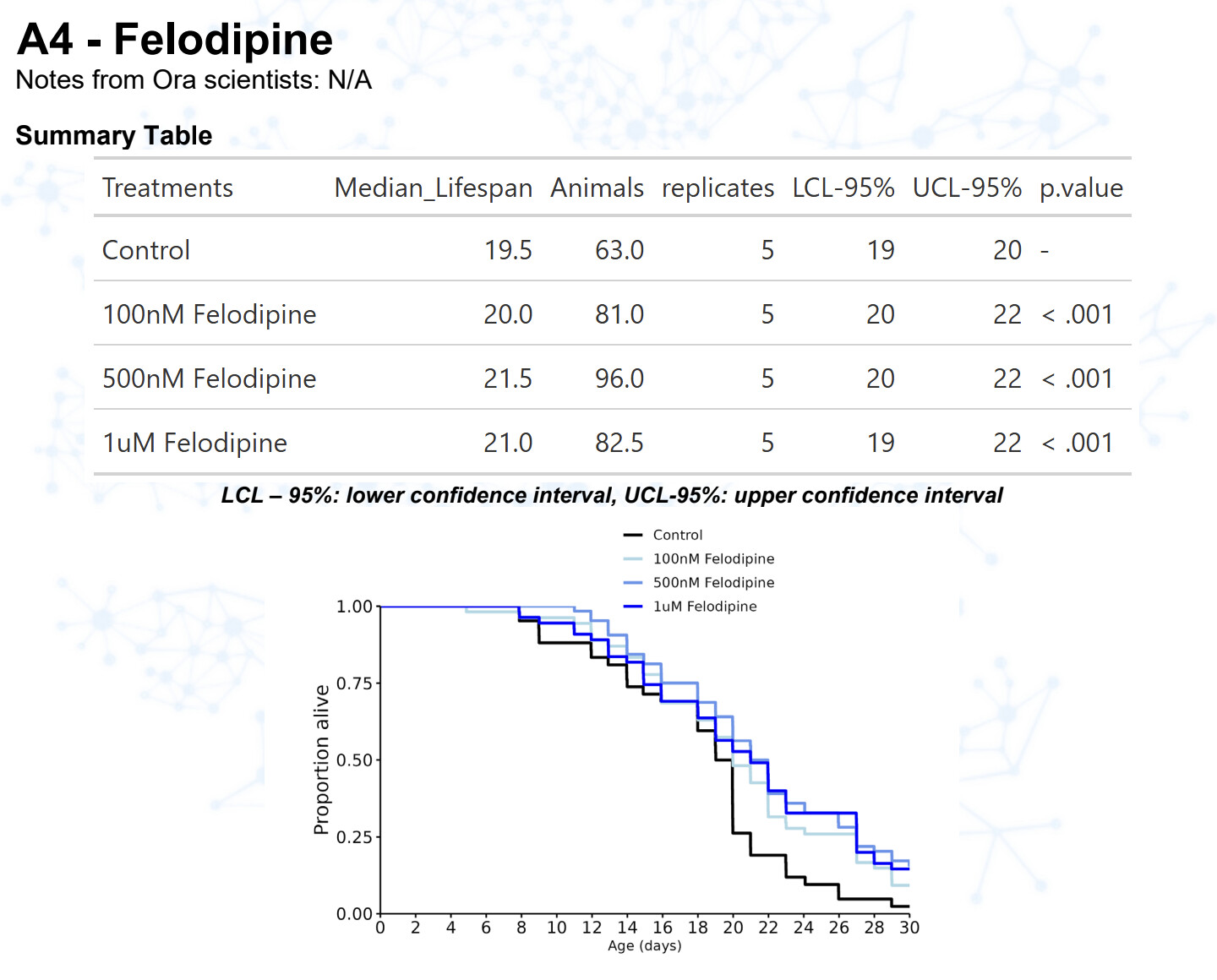
Amlodipine
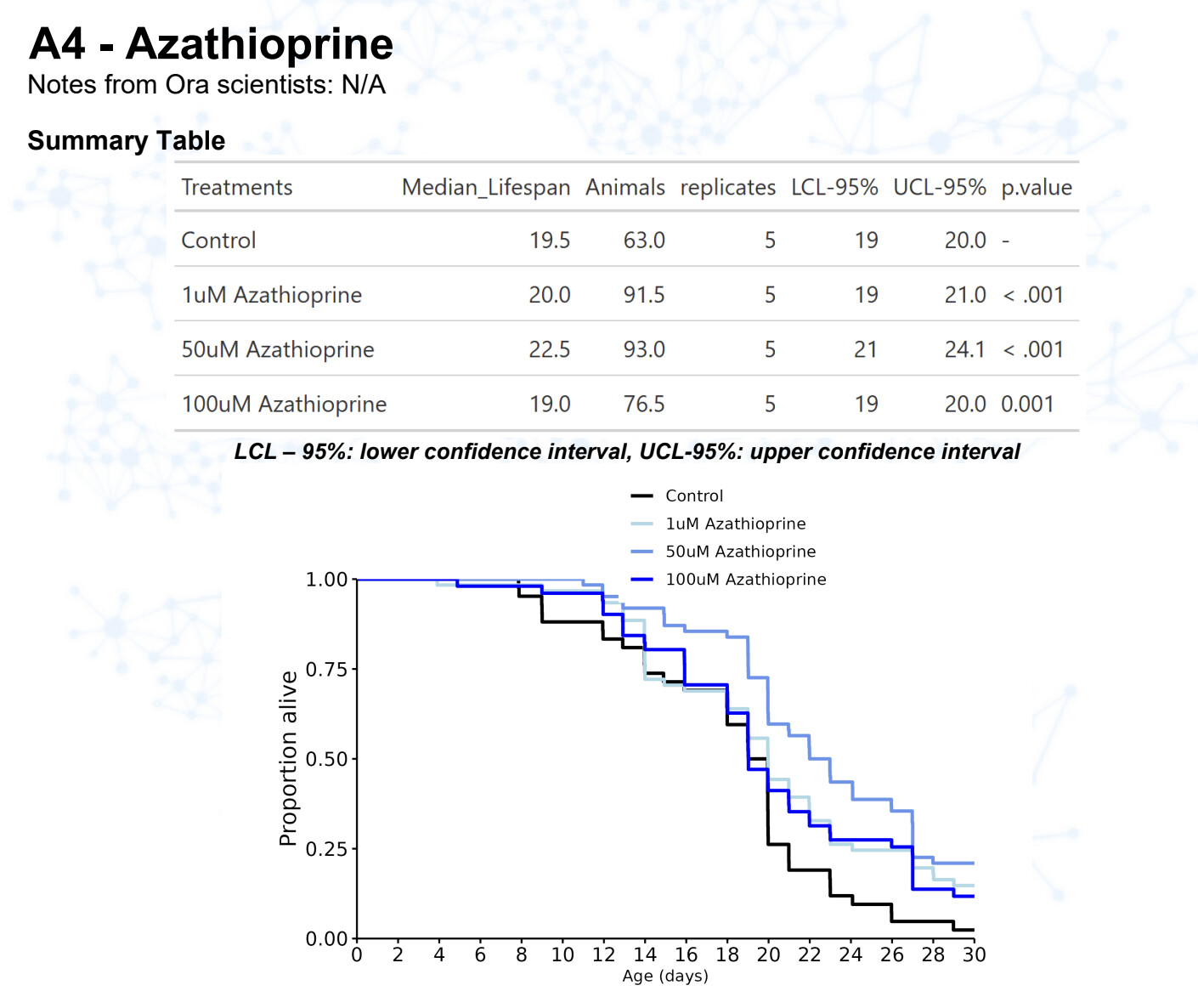
Azathioprine
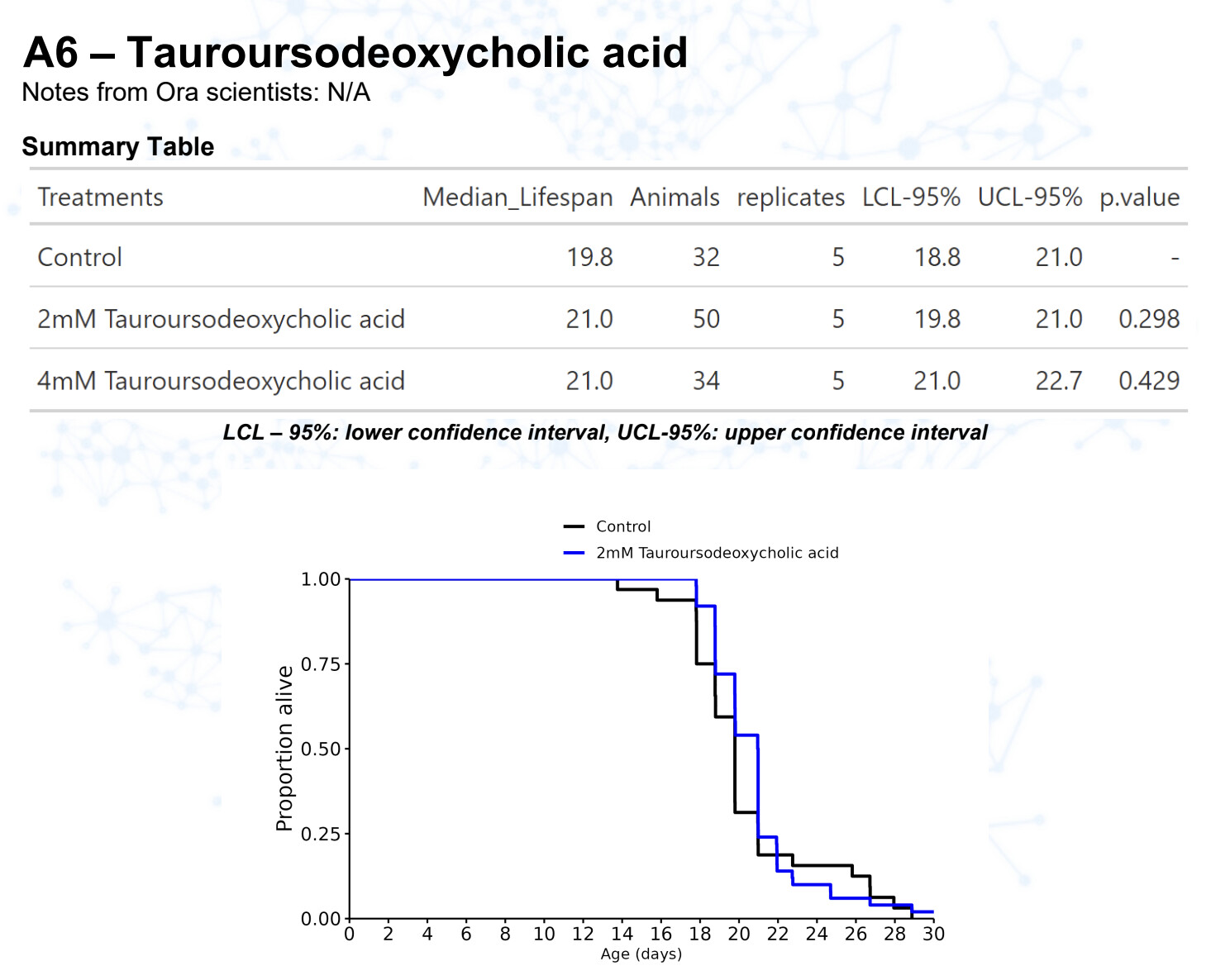
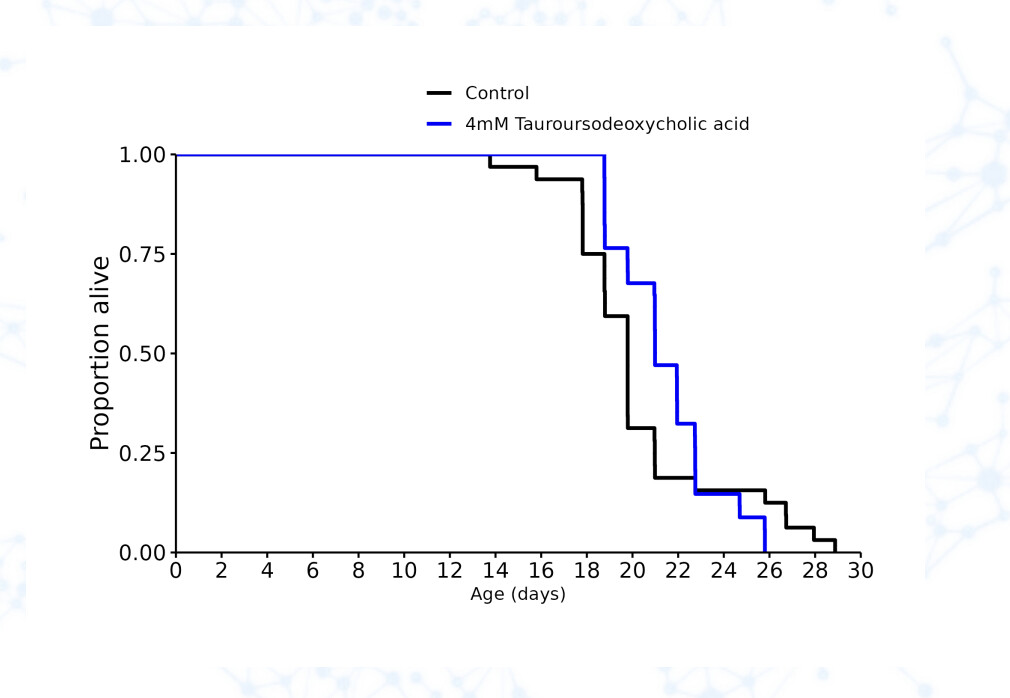
TUDCA
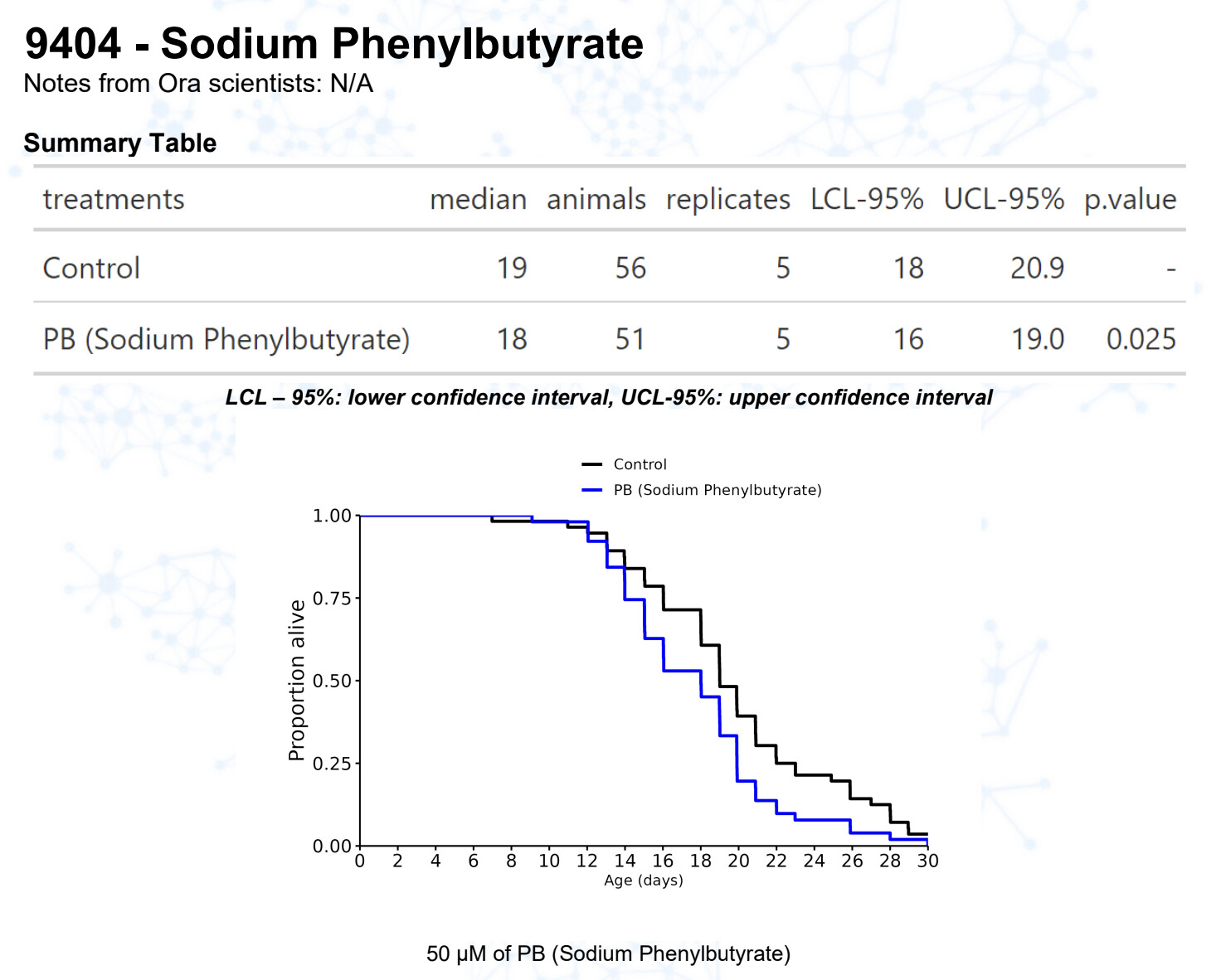
Sodium Phenylbutyrate
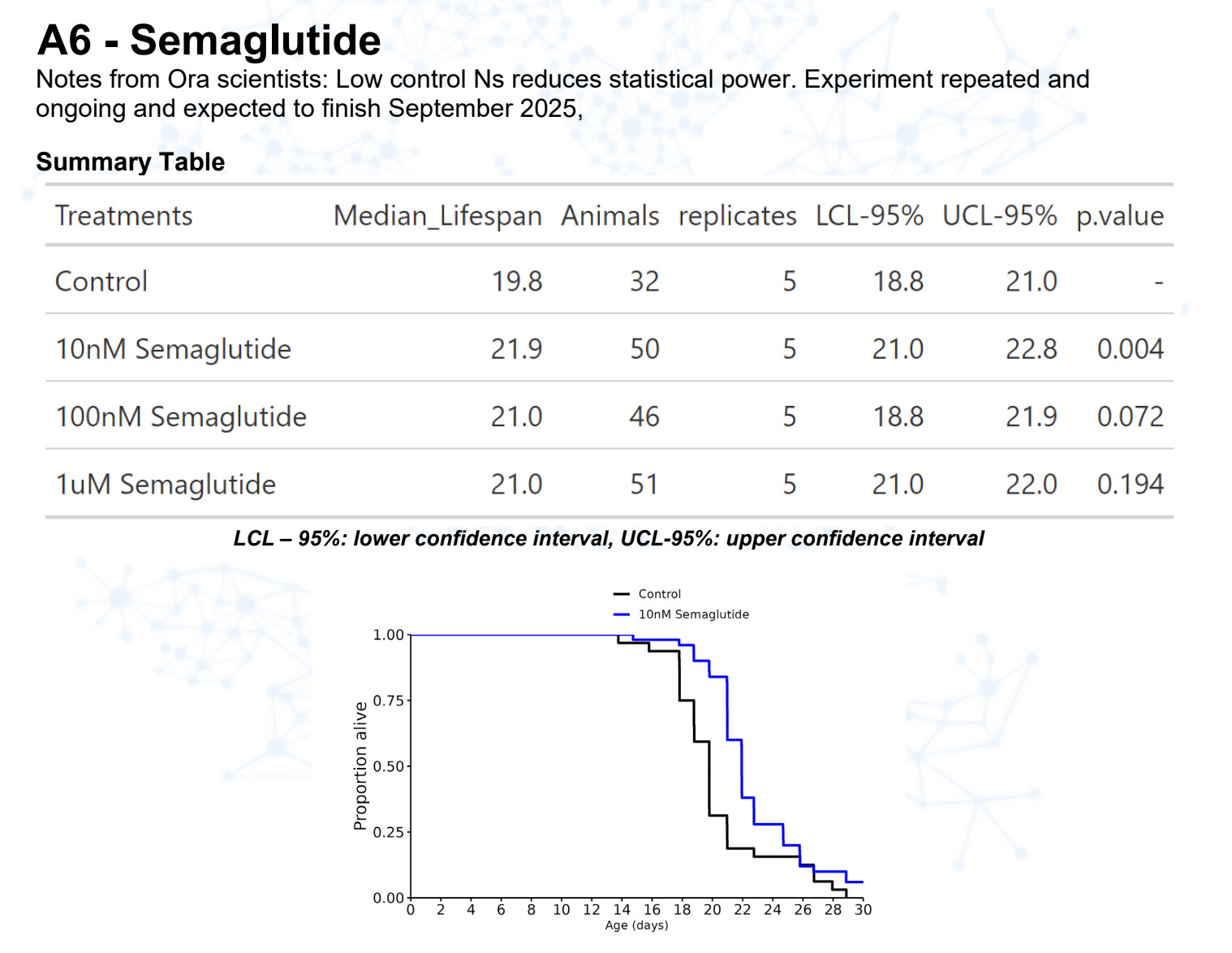
Semaglutide
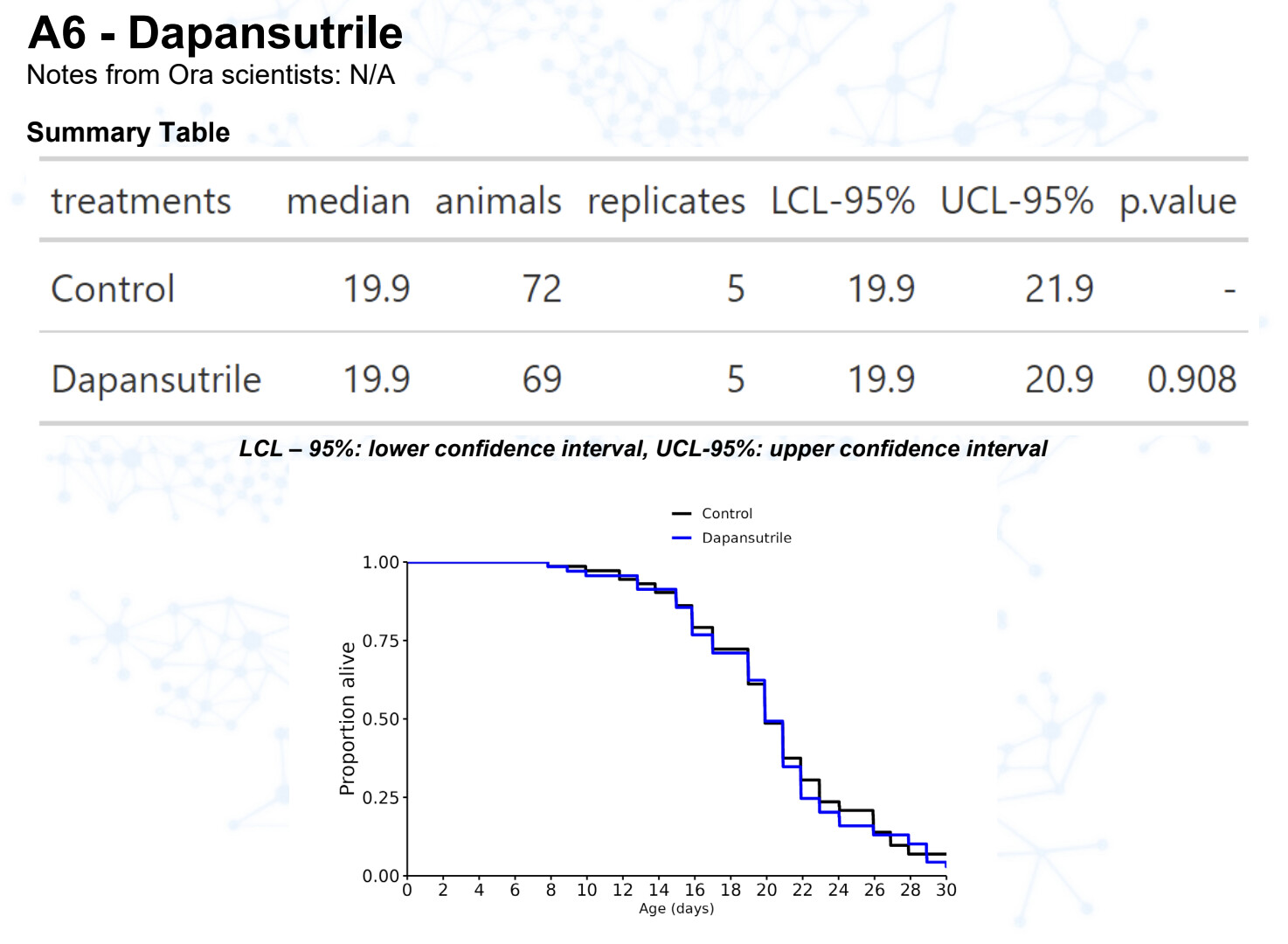
Dapansutrile
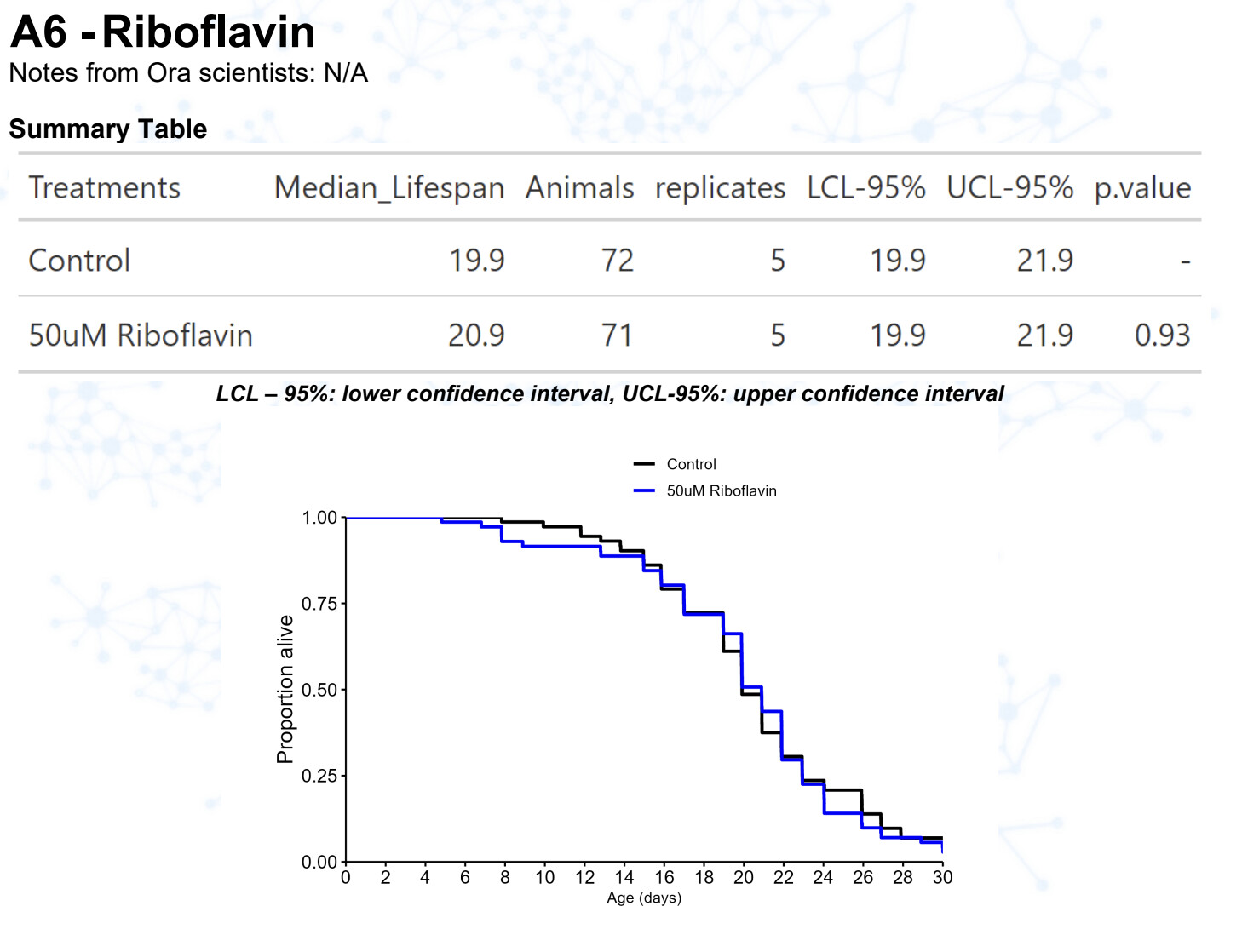
Riboflavin (vitamin B2)
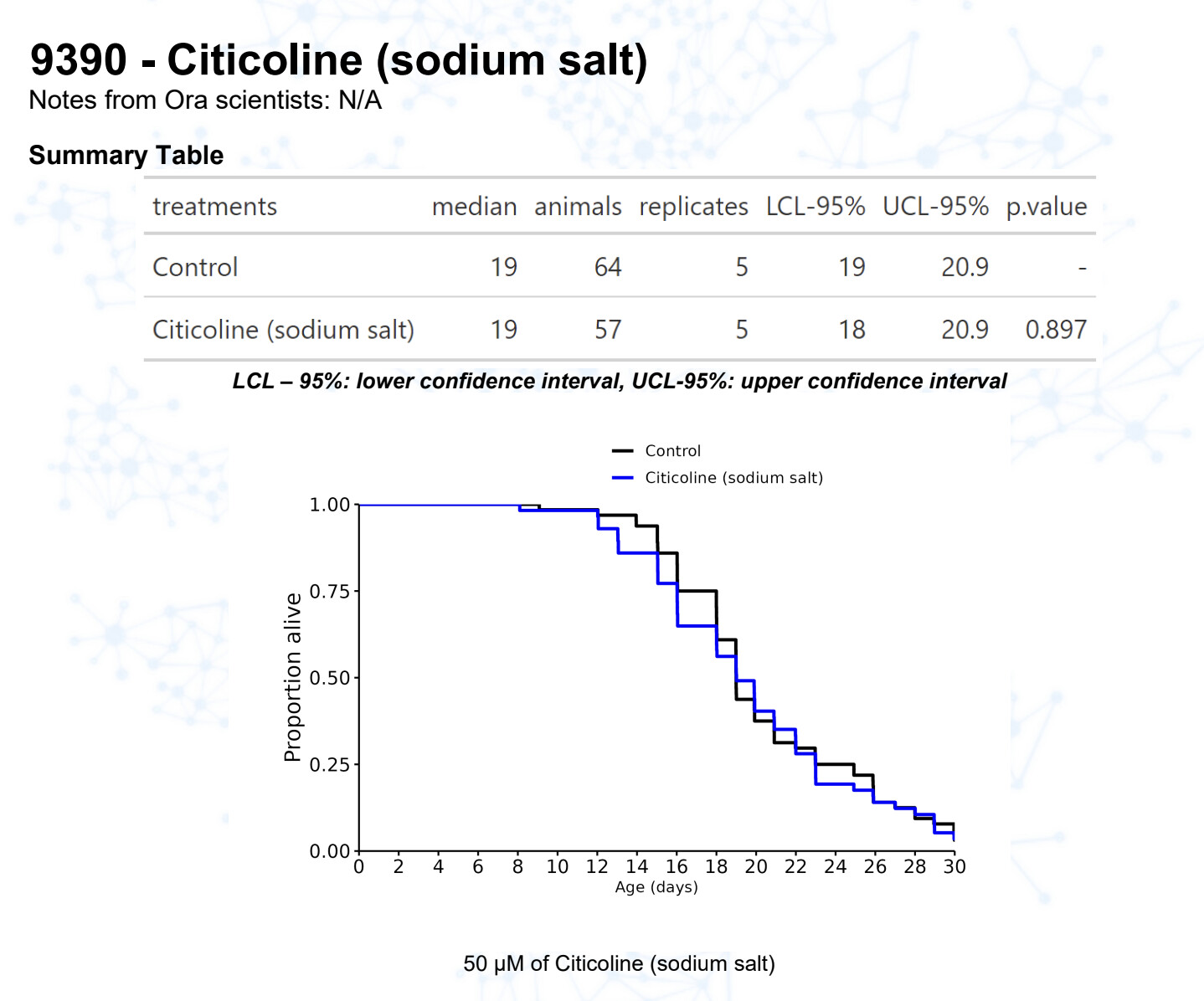
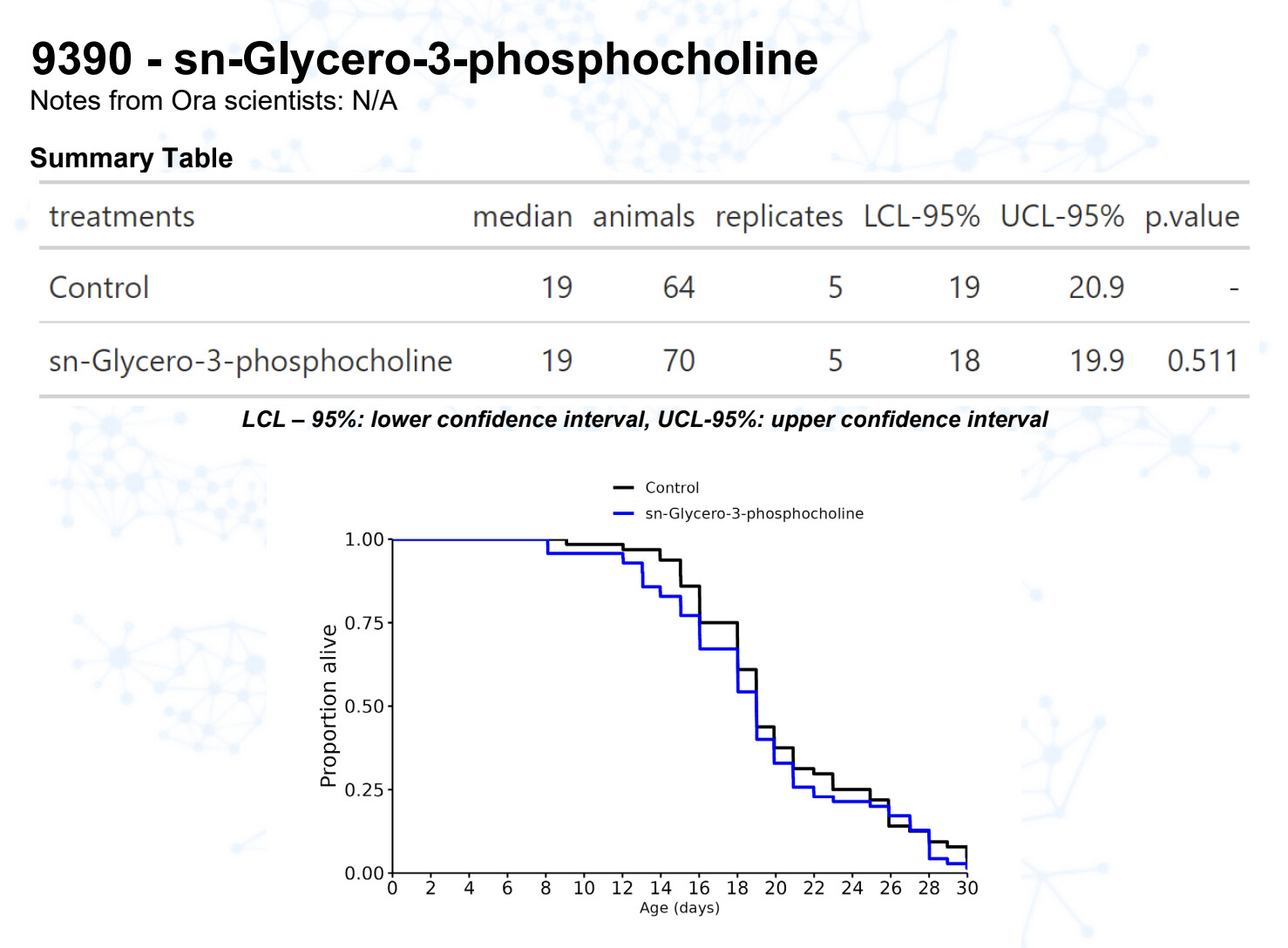
Citicoline
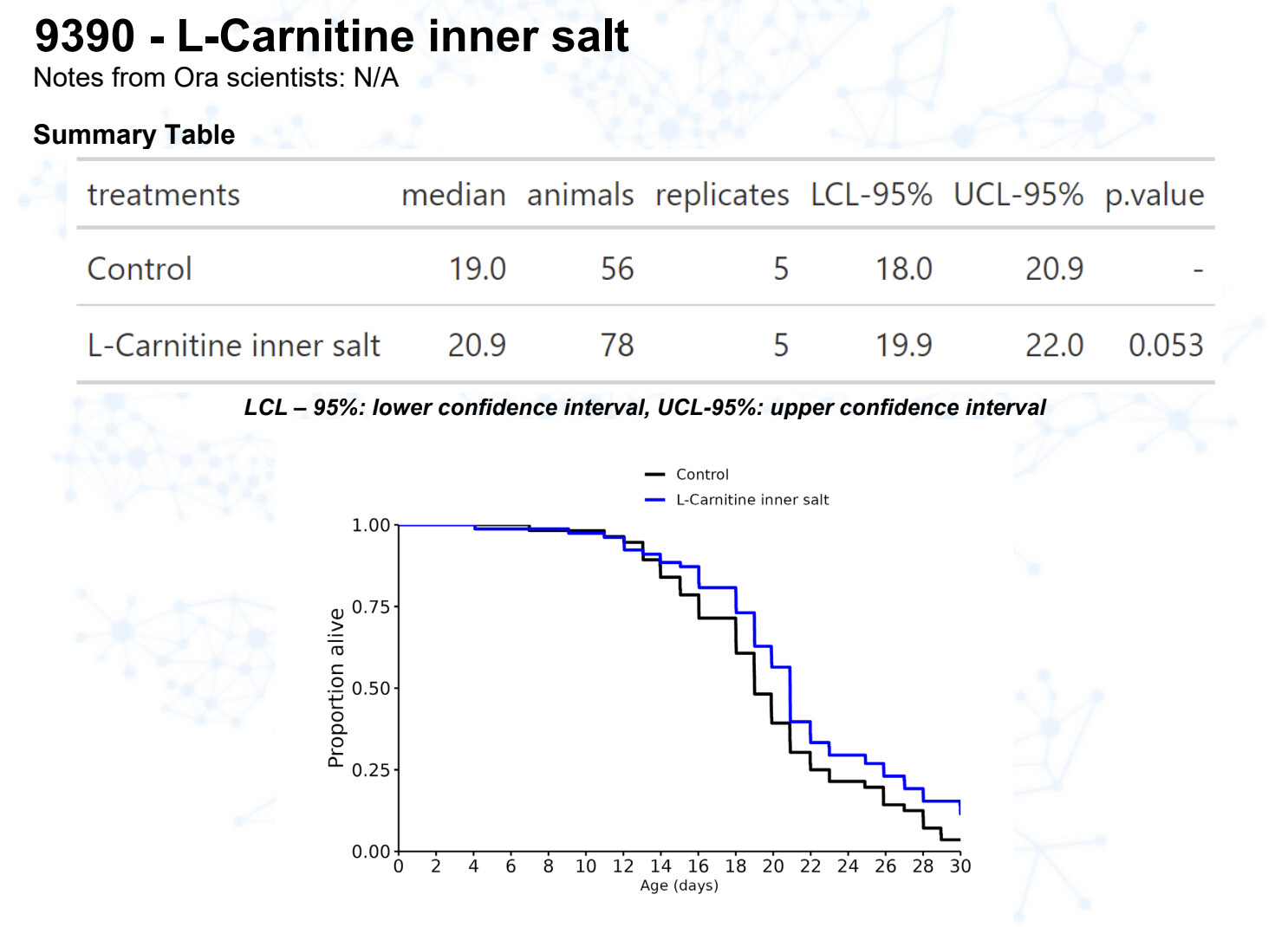
L-carnitine
Alpha-GPC
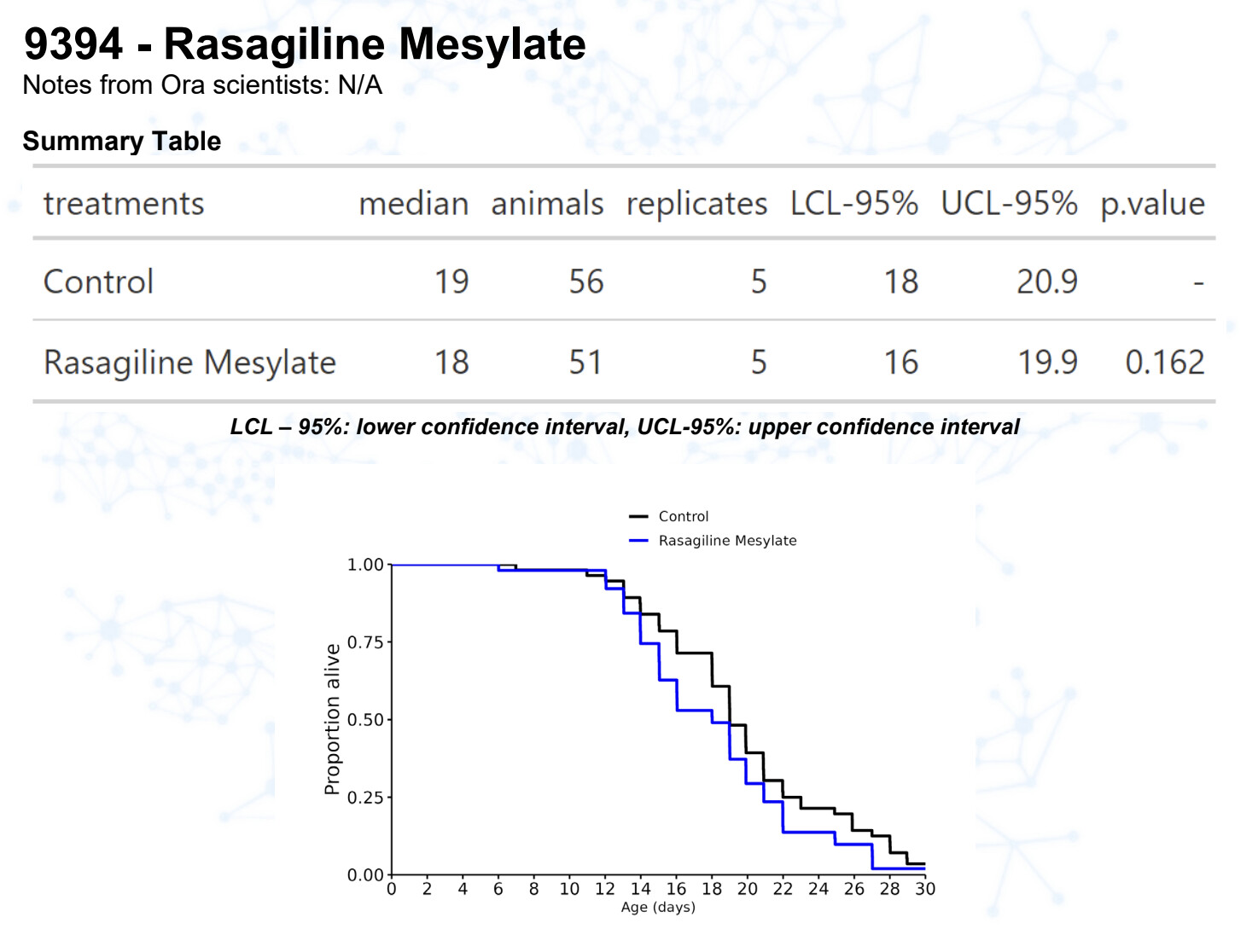
Rasagiline M
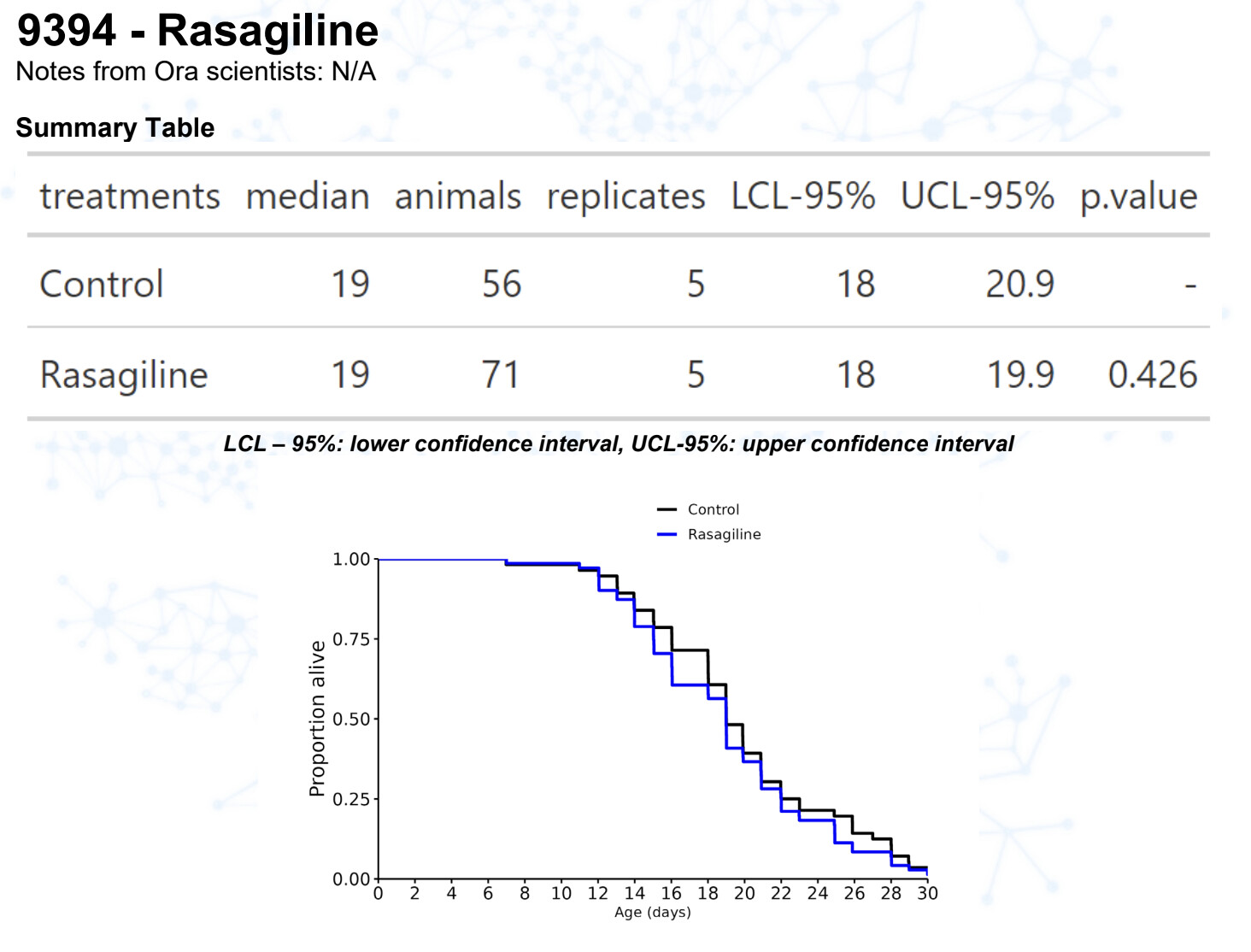
Rasagiline
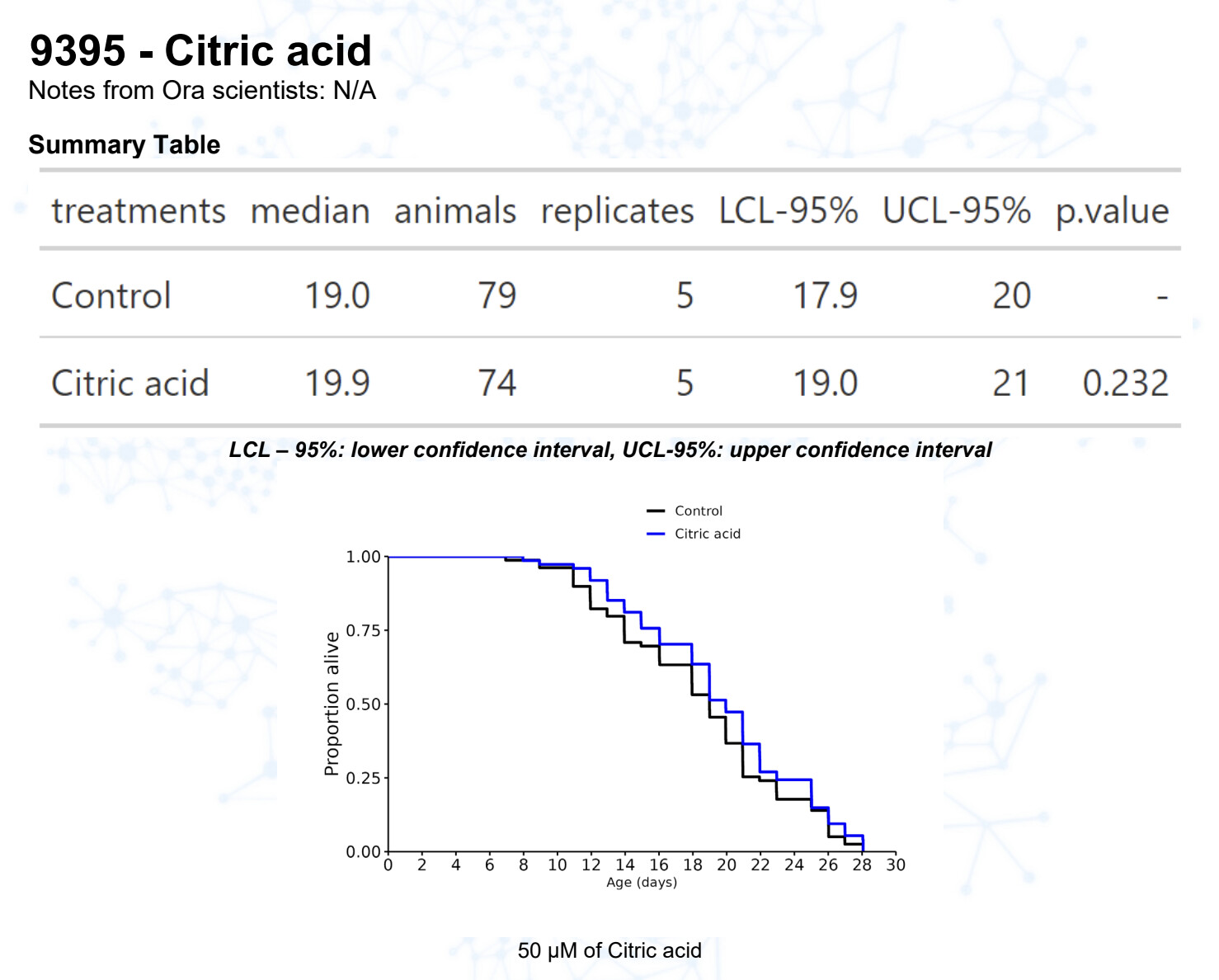
Citric acid (@John_Hemming)
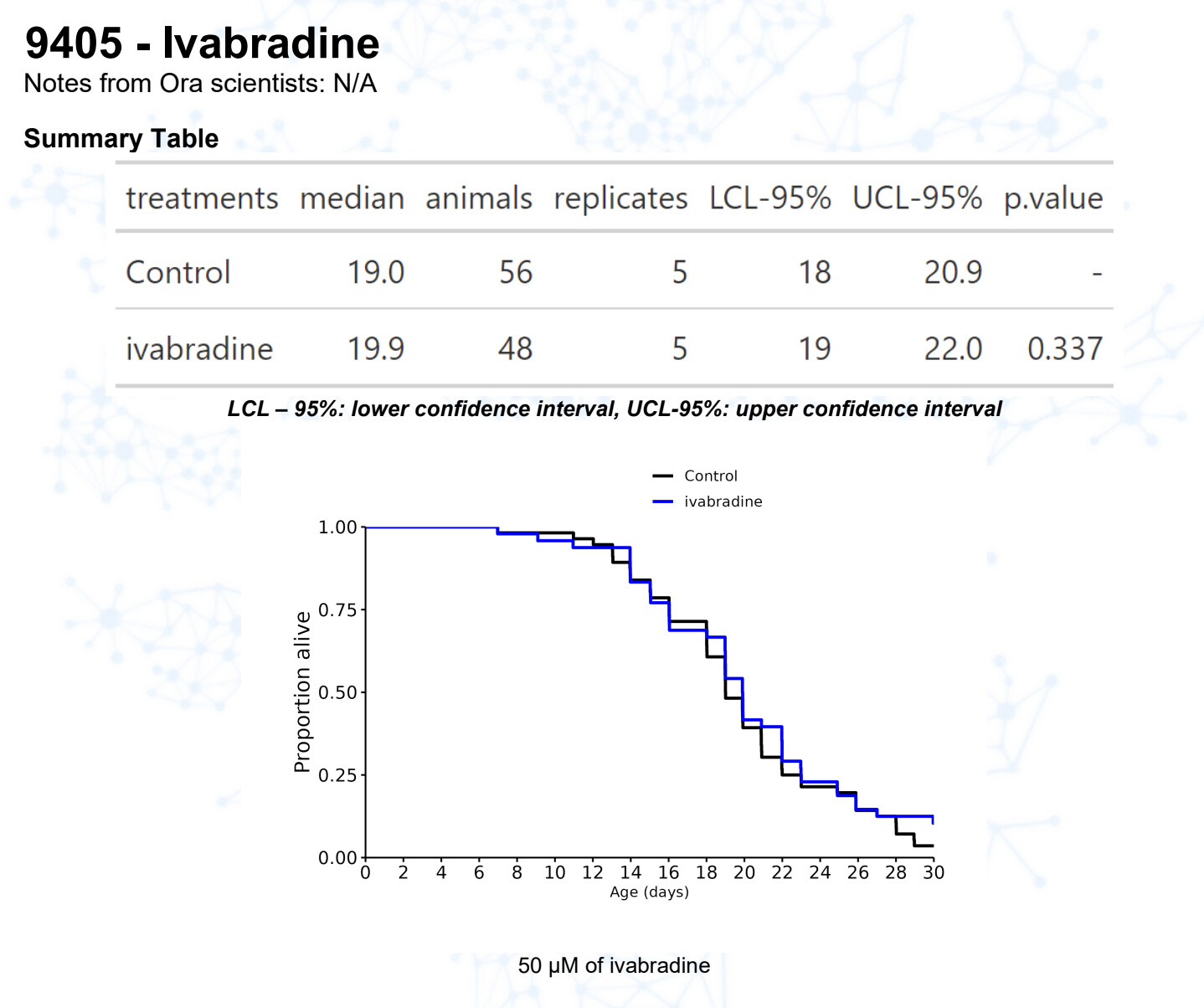
Ivabradine
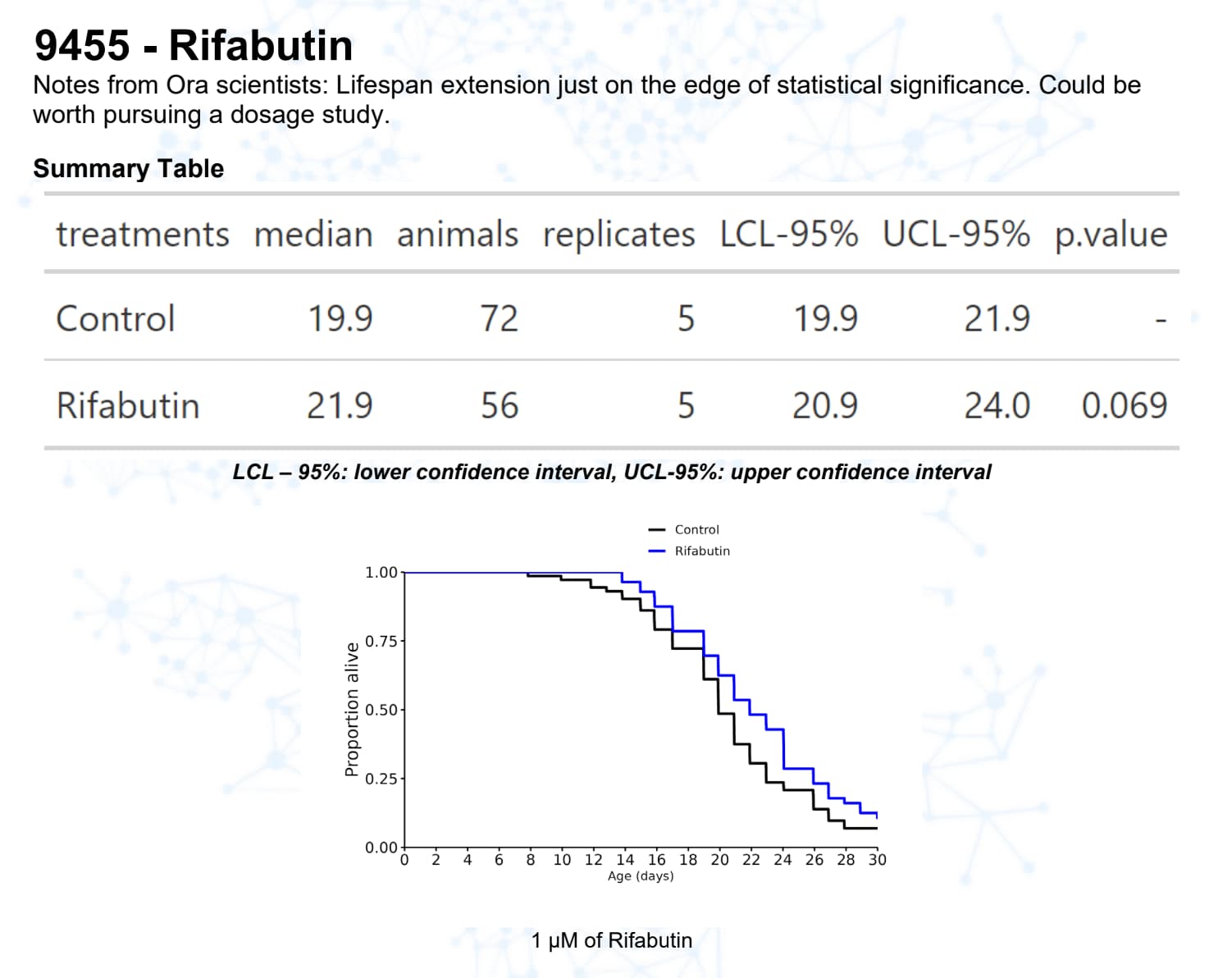
Rifabutin
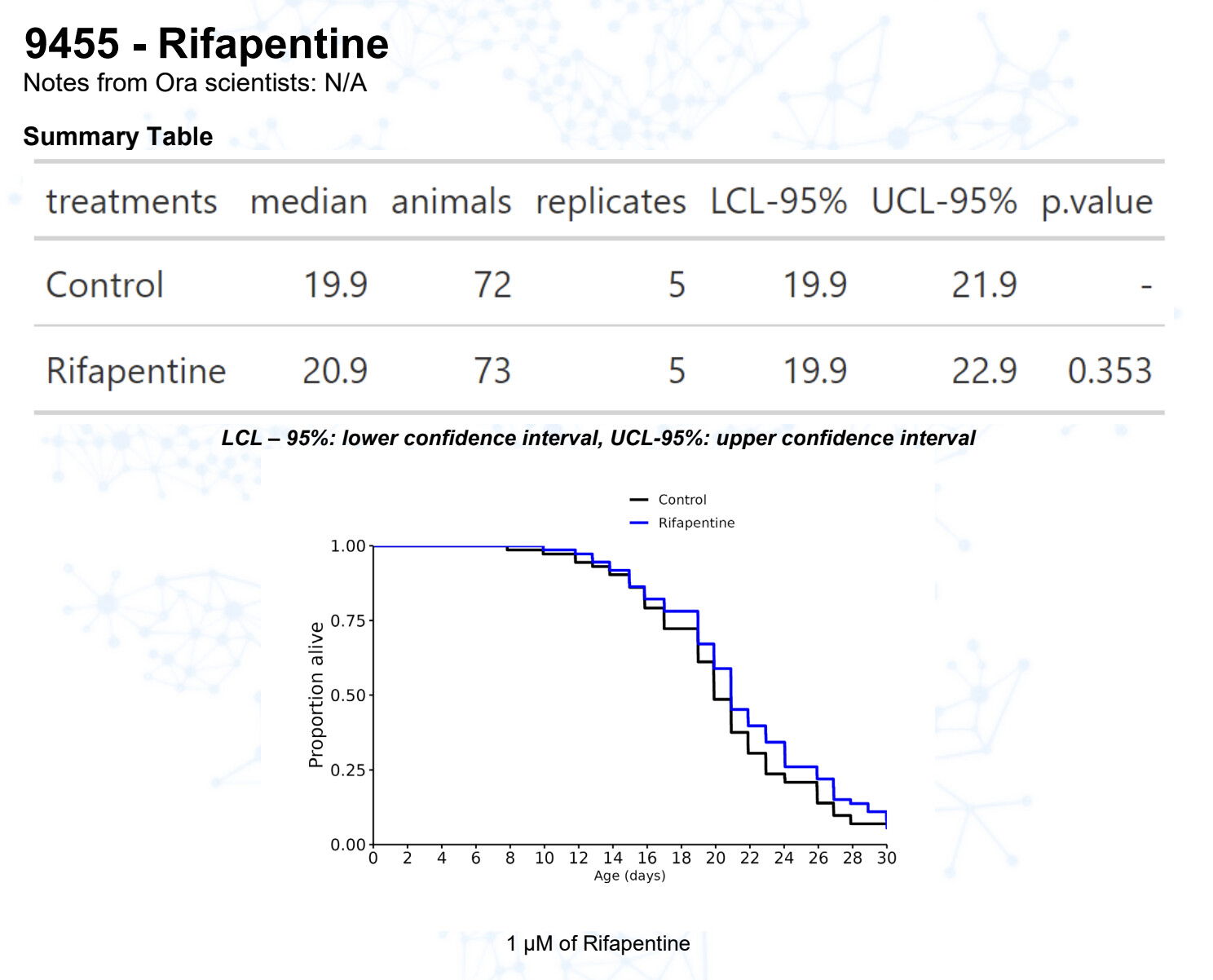
Rifapentin
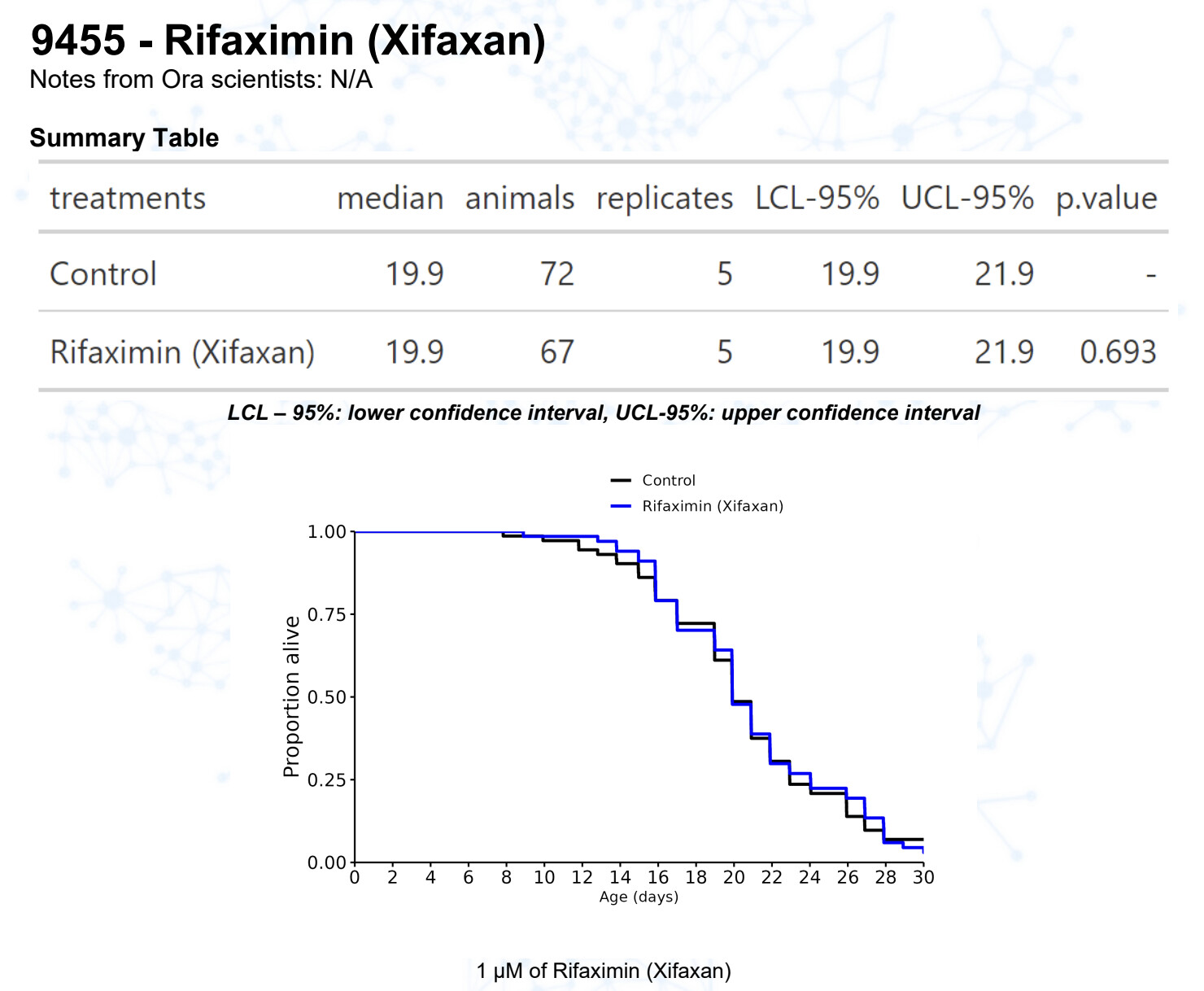
Rifaximin
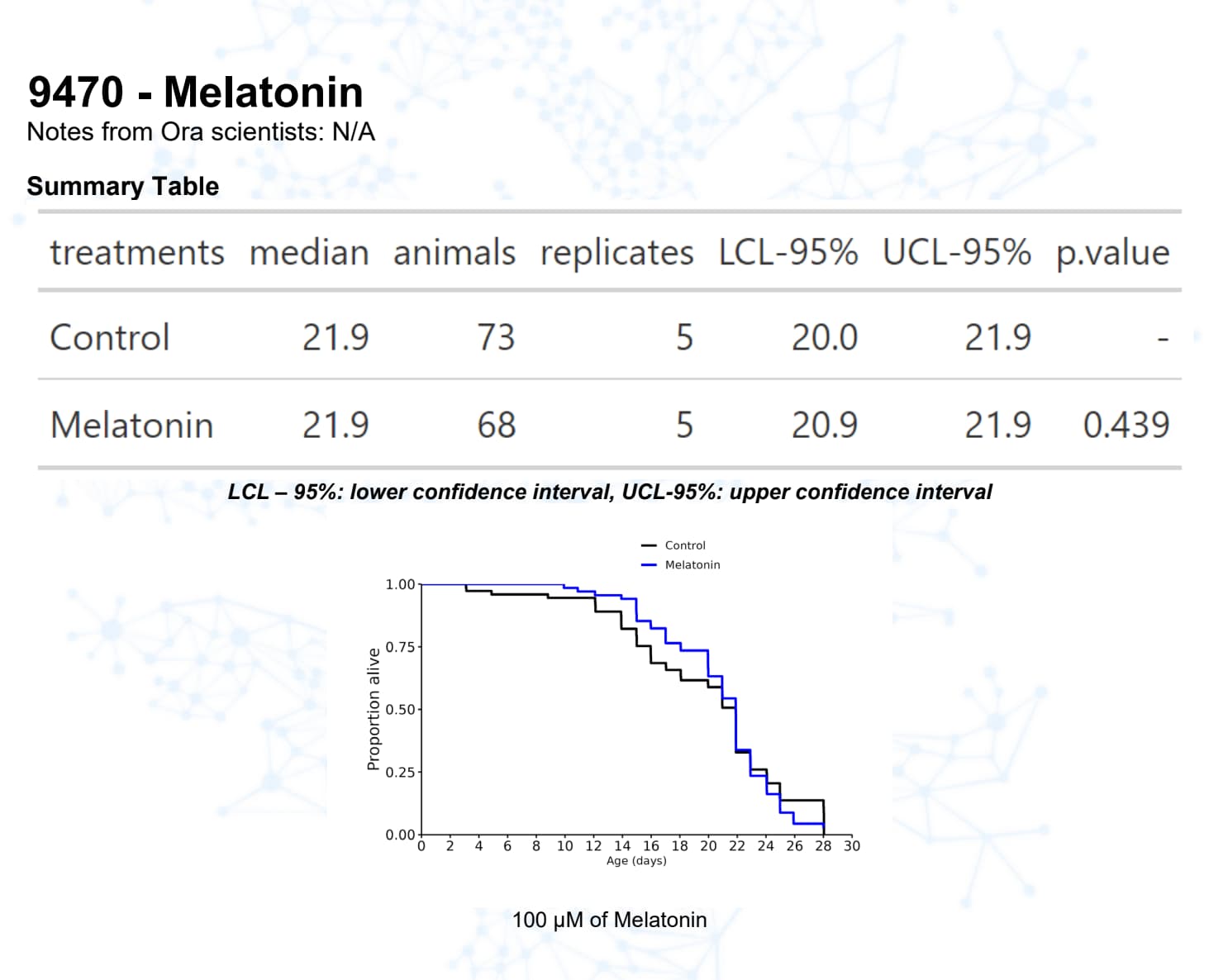
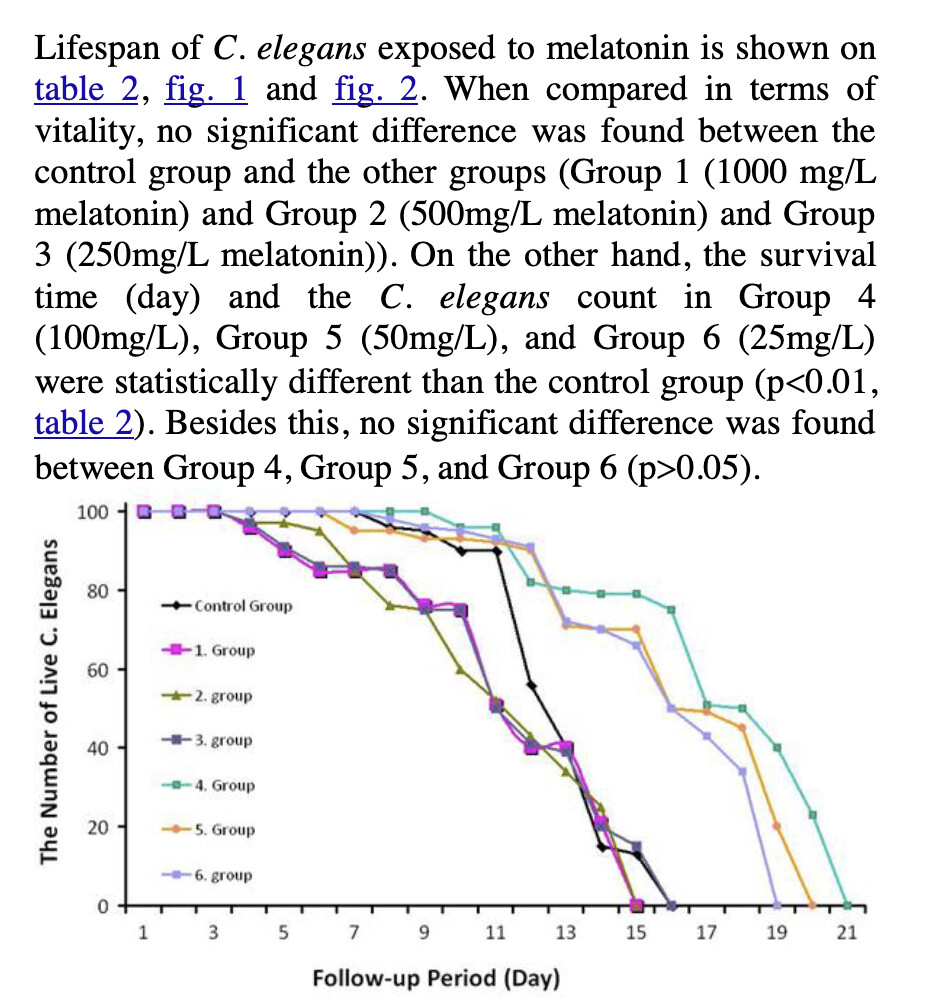
Melatonin
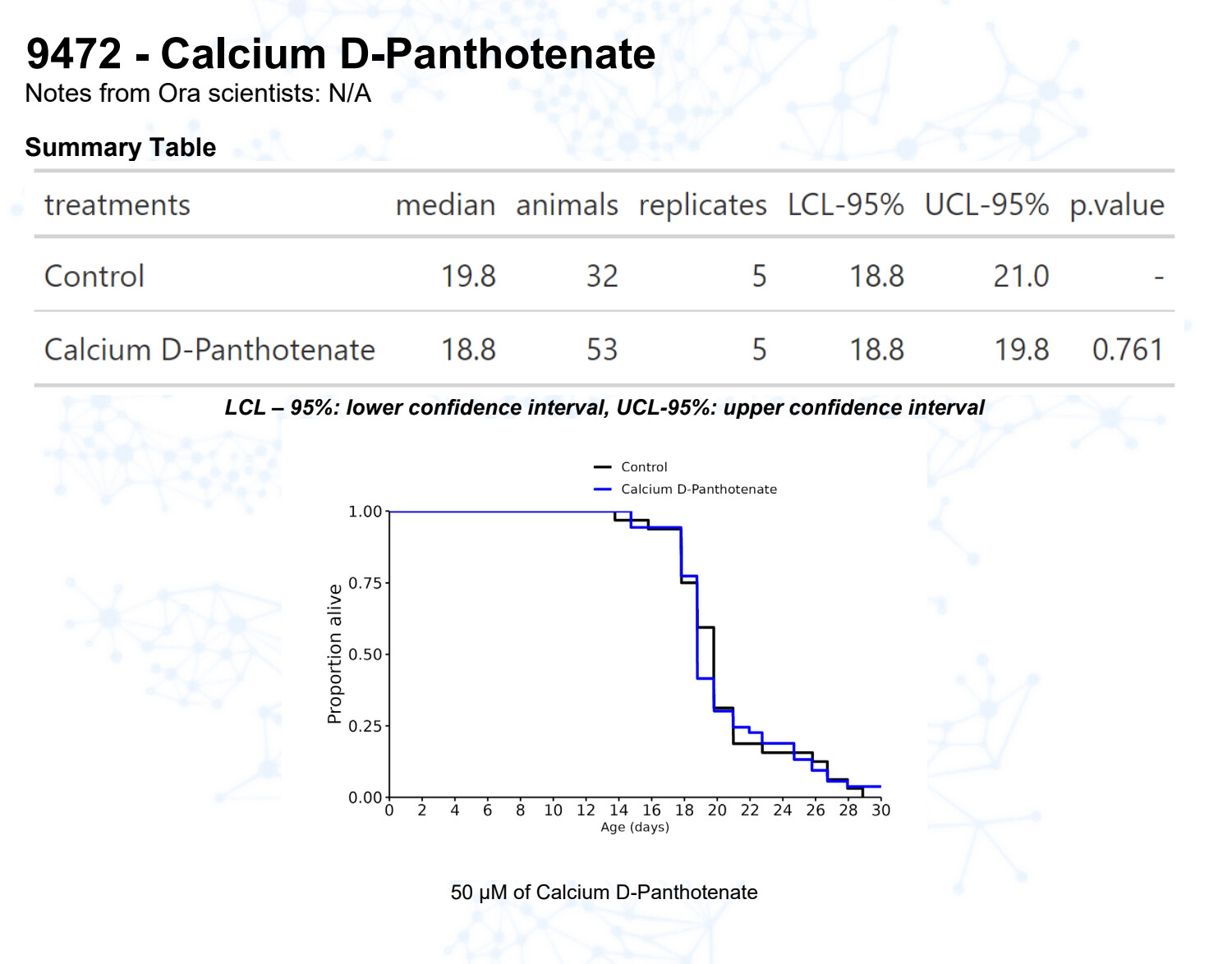
Calcium pantothenate (vitamin B5)
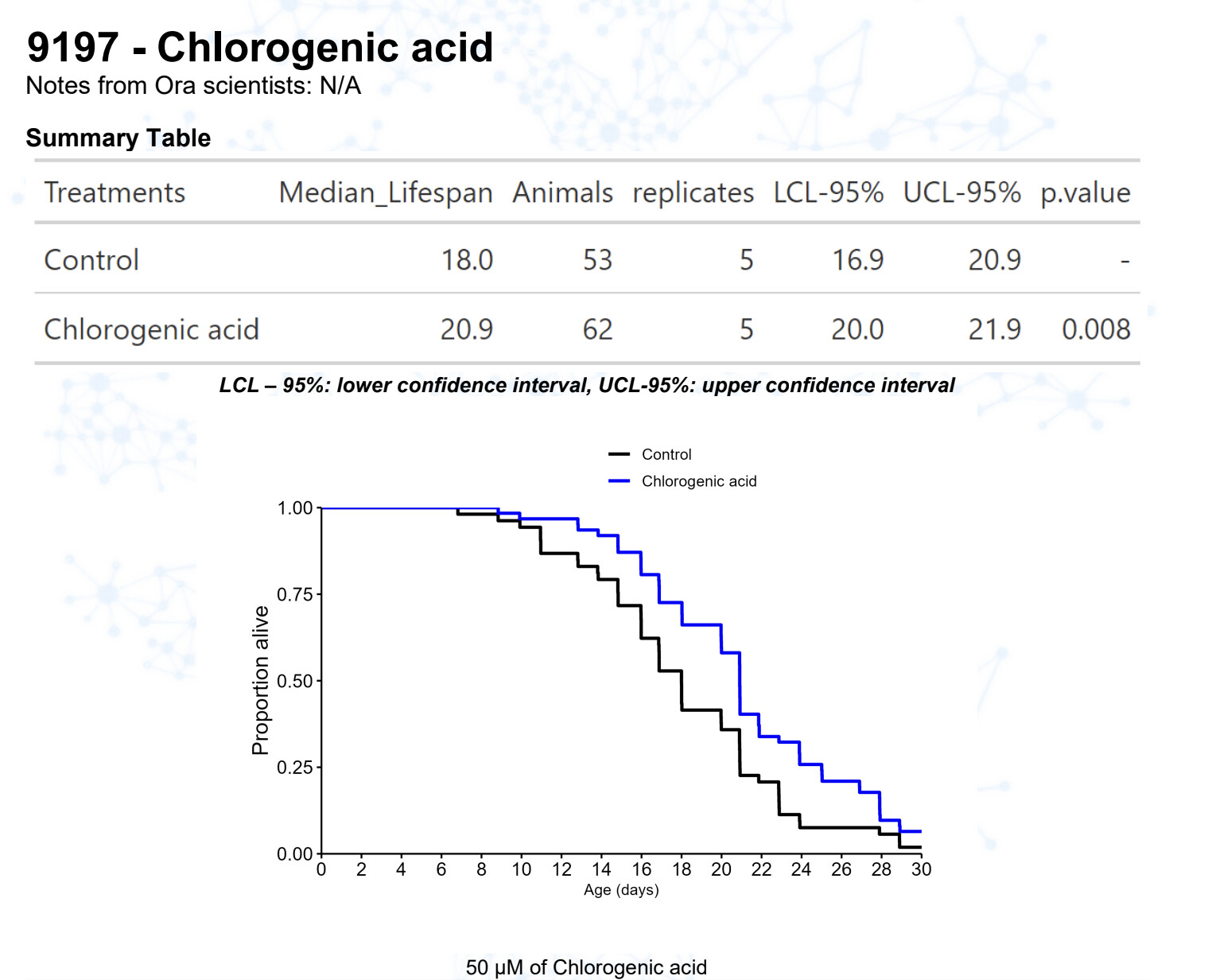
Chlorogenic acid
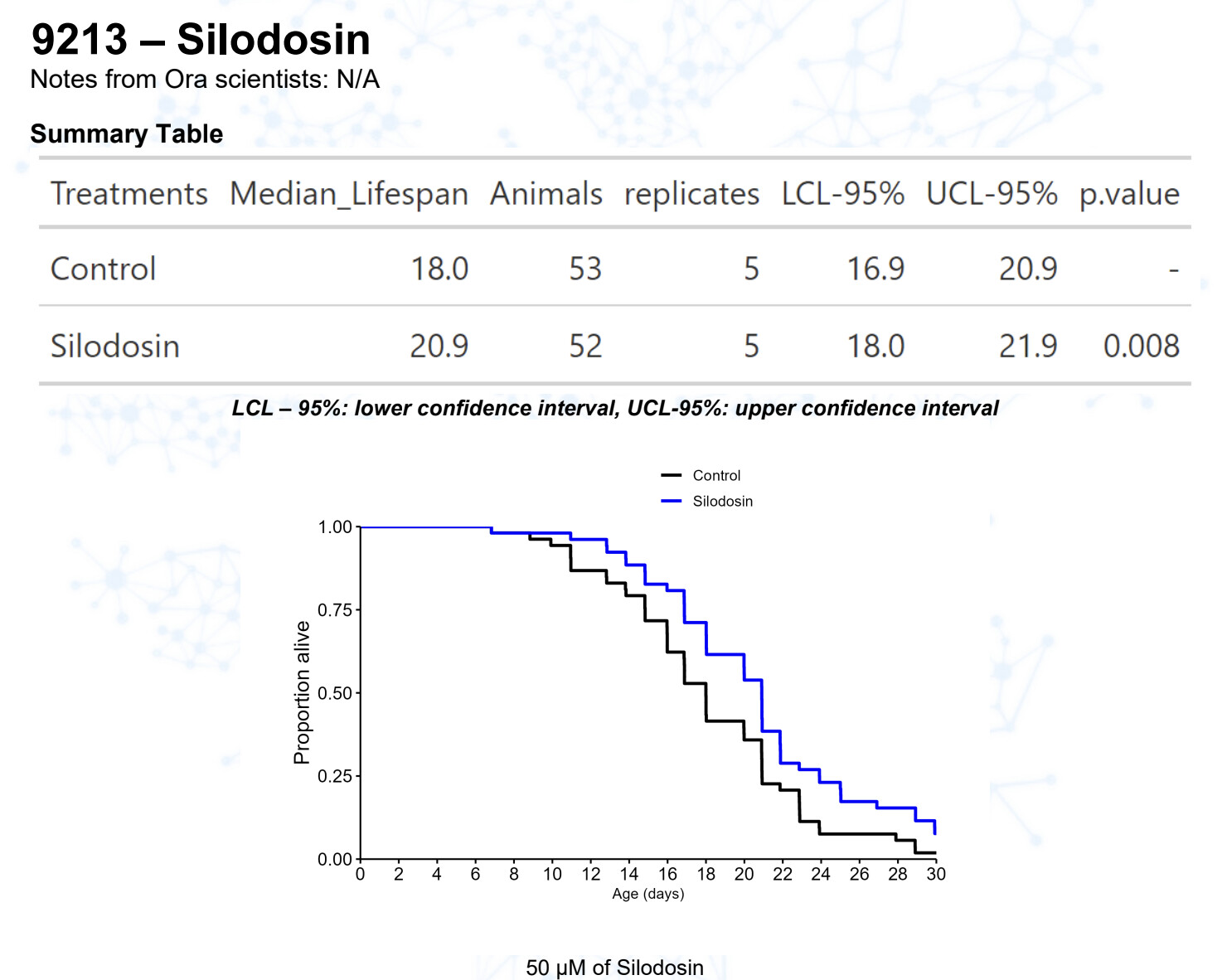
Silodosin
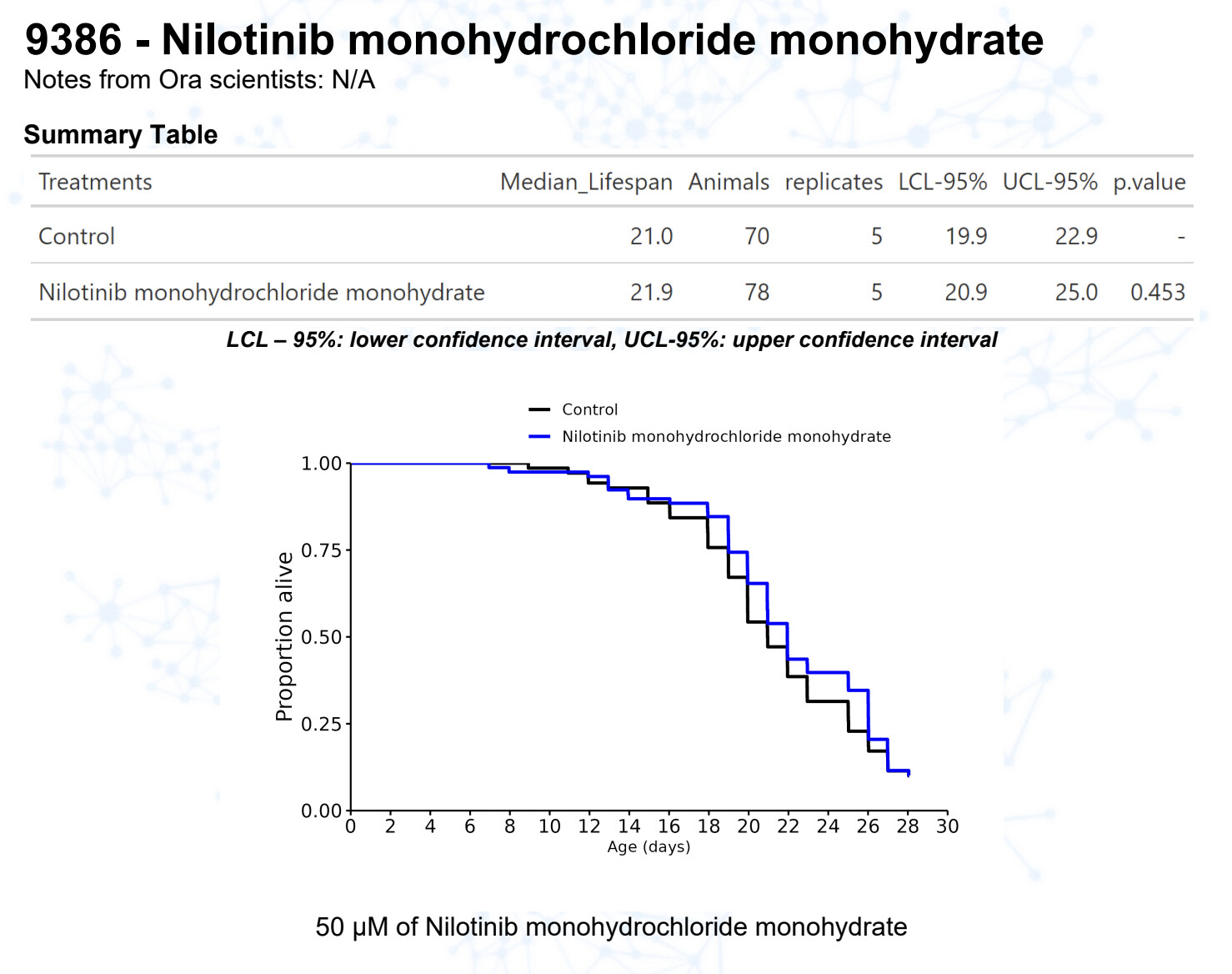
Nilotinib monohydrochloride monohydrate
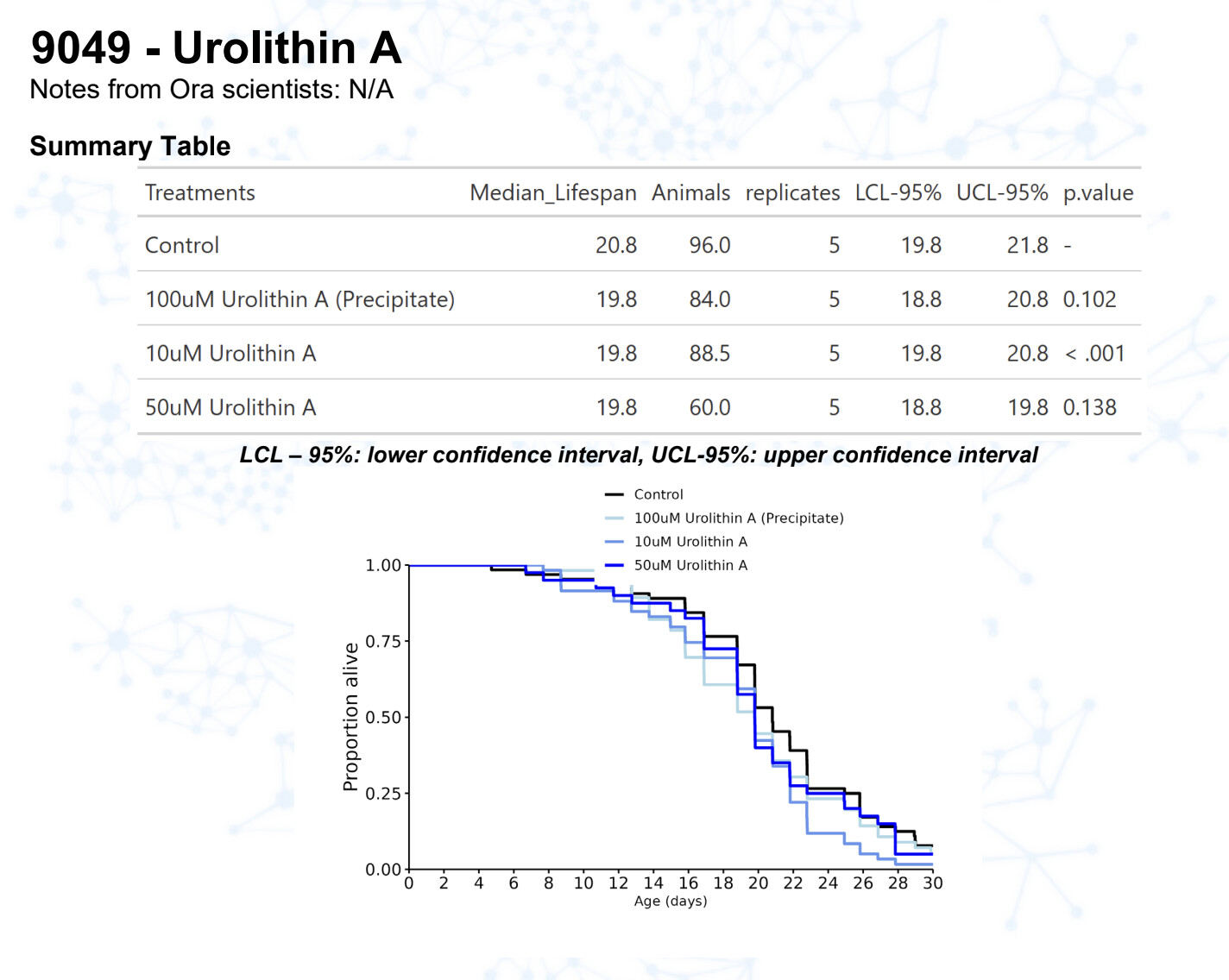
Urolithin A
15 Likes
adssx
#414
I’m planning to test next:
12 Likes
Some eyebrow raisers. Urolithin A, lol. I take citicoline, hopefully I’m not a worm  .
.
3 Likes
Gpt5: Here you go—concise side-by-side:
| Intervention (class) |
Mammal (mouse) lifespan evidence |
C. elegans result |
Notes / why it differs |
| 17-α-estradiol (non-feminizing estrogen) |
Extends male UM-HET3 mouse lifespan; benefits even when started late in life. |
CITP multi-site testing found it broadly ineffectual across Caenorhabditis strains (one strain exception only). |
Likely sex-specific metabolic/endocrine mechanism in mammals that doesn’t translate cleanly to worms. |
| Acarbose (α-glucosidase inhibitor) |
Robust lifespan extension, especially in males, across multiple doses in ITP mice. |
No significant lifespan change in any tested Caenorhabditis strain (CITP). |
Gut/post-prandial glucose dynamics and microbiome–host interactions differ from worms’ simple gut physiology. |
| Canagliflozin (SGLT2 inhibitor) |
Extends male mouse median lifespan by ~14% (ITP). |
No demonstrated worm lifespan benefit; worms lack characterized SGLT transporters—only a GLUT-like transporter (FGT-1) is identified. |
Target (SGLT2) not conserved in C. elegans intestine; mechanism unlikely to apply. |
| Senescent cell clearance (genetic INK-ATTAC; senolytics like dasatinib+quercetin) |
Genetic clearance of p16Ink4a⁺ cells extends lifespan; senolytics improve late-life survival/health. |
Adult worm soma is largely post-mitotic and does not exhibit Hayflick-type cellular senescence, so there’s no comparable target. |
Mechanism is mammal-specific; worms age without proliferative cellular senescence. |
| Growth-hormone axis manipulations (Ames/Snell dwarf; GHR/GHRH knockouts) |
Large lifespan gains (often 40–60%) in dwarf/GH-axis mutant mice; GHR/GHRH disruption extends lifespan. |
No GH axis in nematodes (GH evolved in vertebrates); worms instead use insulin-like signaling for growth/aging. |
Endocrine lever is vertebrate-specific, so it doesn’t exist in C. elegans. |
If you want this exported (CSV/Excel) or expanded with effect sizes and dosing, say the word and I’ll generate it.
6 Likes
adssx
#418
What’s weird is that a paper found 50% life extension in worms: https://onlinelibrary.wiley.com/doi/10.1111/acel.14424
I’ll ask Ora why such a discrepancy.
Also (as I think John is alluding to): null results are not that significant. But we should care about a significant life extension or shortening and move these compounds to larger animal models.
7 Likes
a) What has been found to extend life in mammals but not in c elegans
then
b) Do table
I also worry personally about the food question. Hence something that helps E Coli can harm C Elegans lifespan. SImilarly if you kill E coli you extend C Elegans life.
3 Likes
What I think is that the experiments are easy, fast and cheap to do. As a general principle things that are easy, fast and cheap are not good quality and the information we get is not good quality. It is, however, better than having no information at all.
Melatonin, for example, has a good track record in mice.
Citrate, has a good track record in drosophila.
If I get a bit of time I intend reading up on the difference between mtDNA mutations in C Elegans and other species.
I think the relative absence of PINK1/Parkin is probably a key element in the short lifespan.
1 Like
adssx
#421
2 Likes