adssx
#484
Tiny (n=15 in the nicotine group) short (12 weeks) Iranian trial without washout period = trash.
Also, what is a “nicotine-rich diet”? 
A serious larger study found WORSENING of symptoms on nicotine:
The study showed no benefit of nicotine, and in fact a trend was observed toward an accelerated decline in function in the nicotine group, with UPDRS parts I–III score increased 3.5 points in the placebo arm (N = 54) versus 6.0 points in the nicotine arm (N = 47, P = 0.056) over the 60 weeks. Secondary analysis of the same measure over the full treatment period but prior to washout (ie, over 52 weeks) on a larger portion of the cohort (N=138) also showed greater worsening on nicotine compared to placebo (P = 0.010).
See:
2 Likes
Davin8r
#485
Tobacco leaf salad with balsamic vinaigrette?
3 Likes
Your bio still says: “Are you using Rapamycin now?: Not Yet - But Interested”
Is this still true?
FWIW:
When I first started taking rapamycin, I had “age-related essential tremors”,
This mainly manifested in shaky hands.
After taking high-dose rapamycin(up to 20 mg weekly) for the first few weeks, they completely disappeared and haven’t returned.
“Age-related essential tremors (ART) and Parkinson’s disease (PD) are distinct clinical entities, but there is evidence suggesting a potential relationship between them.”
8 Likes
adssx
#487
That’s valuable feedback, thanks!
I don’t use rapamycin because:
- It does not seem to cross the blood-brain barrier
- Anecdotes of people with PD who tried it (such as @TomParkinson here) don’t report benefits
- It failed in a clinical trial in MSA (a condition close to PD)
- Research is not concluant
- It might mess up with glycemic control and mine is already suboptimal
That’s why I looked at everolimus: Everolimus instead of Sirolimus / Rapamycin? Anyone else trying? - #177 by CronosTempi
However, the evidence in favor of everolimus is not much better. So I gave up.
Still, it might be worth giving a try to sirolimus (I was thinking about it this morning, so your message arrives at the perfect time!). Especially if, as in your case, the benefits appear quickly. Which dose did you start with? How many weeks for the tremors to disappear?
I’m testing a few other things now so I’ll wait a bit to test rapa. Also, a trial of another immunosuppressant, azathioprine, has just ended. We should have the results in April. I heard they will be “interesting”. That might strengthen the case for rapa?
5 Likes
A_User
#488
I don’t know if you’ve seen this paper (there’s so many papers & discussions).
There are a few threads on ketamine & rapamycin and videos from the lead author of the experiment which showed rapa prolonging the anti-depressant effect (they tried it because they believed it would block it).
From the paper it seems that it does cross the BBB:
Using a randomized placebo-controlled cross-over design, rapamycin was administered as a single 6 mg dose prior to ketamine infusion. In several species, preclinical studies have shown that rapamycin crosses the blood brain barrier, as measured by rapamycin levels in the cerebrospinal fluid and brain tissues, or by the inhibition of brain mTORC1 signaling [23–26]. Moreover, within 2 h following peripheral rapamycin administration, one study reported decreased phosphorylation of S6 ribosomal protein in brain tissues—a pharmacodynamic readout of mTORC1 inhibition [15]. Furthermore, the immunosuppressive effect of rapamycin is an mTORC1-dependent process [27] and rapamycin was shown at therapeutic doses in humans to cross the brain blood barrier and to reduce the phosphorylation of S6 ribosomal protein in brain tissue [28, 29]. Therefore, the rapamycin dose and timing were selected based on the drug pharmacokinetics to ensure, at the time of ketamine administration, blood concentration of 5–20 ng/mL, a level that exhibits potent immunosuppression [29]. Consistent with the hypothesized mechanism of action of ketamine, we predicted that rapamycin would reduce the antidepressant effects of ketamine.
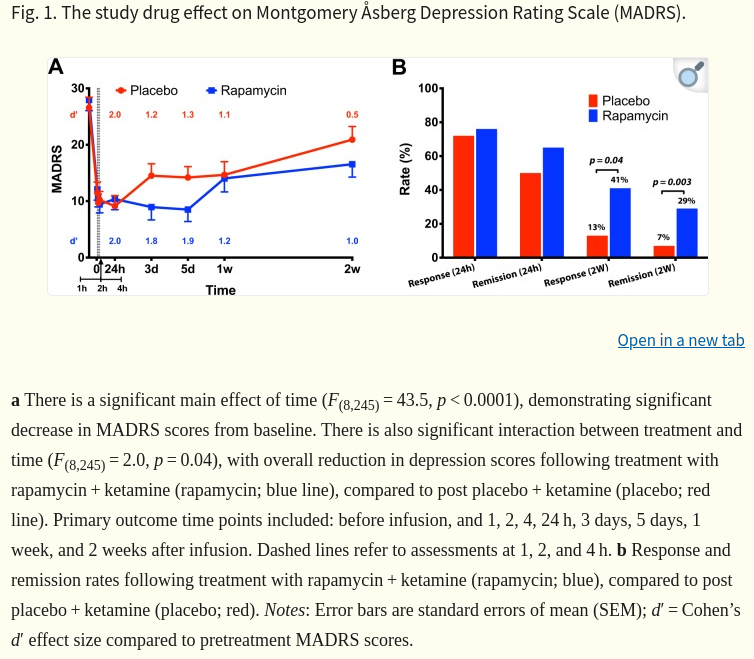
This study yielded two surprising, but potentially important, clinical observations. First, this study failed to validate the prediction from preclinical studies [10, 16], in that rapamycin pretreatment did not reduce the acute antidepressant effects of ketamine at 24 h following treatment. At 24 h, the depression scores and response rates were highly comparable between study arms. Second, rapamycin pretreatment increased the response and remission rates at 2 weeks (Fig. 1b), suggesting that this treatment approach may prolong the antidepressant effects of ketamine. This conclusion is supported by the statistically significant drug by treatment interaction effect on the primary outcome MADRS, showing overall larger reduction in depression scores following rapamycin pretreatment (Fig. 1a). Additionally, the Cohen’s d′ effect size at 2 weeks post rapamycin was 1.0, compared to 0.5 following placebo pretreatment (Fig. 1a). Moreover, the reduction in QIDS-SR scores (secondary outcome) at 2 weeks were significant following rapamycin, but not placebo pretreatment. As well as the response rate using QIDS-SR which was significantly higher at 2 weeks following rapamycin treatment. However, it is important to note the lack of significant difference at 1 week which may indicate a fluctuating course, or it may be related to the relatively small sample size. …
In humans, we are not able to administer rapamycin intracortically to fully parallel the preclinical reports. Further, we limited exposure to rapamycin to a loading immunosuppressant dose, which was selected on the basis of being the highest dose one could administer without exposing subjects to a risk of side effects associated with higher doses [32]. However, we believed that it was important to test whether systemic mTORC1 inhibition blocks the antidepressant effects of ketamine in humans because: (1) The immunosuppressant effects of rapamycin are mTORC1-dependent [27]. (2) There are preclinical and clinical reports providing evidence that peripherally administered rapamycin crosses the blood brain barrier and actively inhibit brain mTORC1 signaling [23–26, 28, 29]. (3) Acute single dose of rapamycin administered peripherally was shown to inhibit mTORC1 in the brain within 2 h of administration in rodents [15]. …
5 Likes
adssx
#489
Thanks a lot you make a very good point I forgot that study!
2 Likes
So sorry to hear about your diagnosis. I have learned much from you on these threads and greatly appreciate all you are doing to share knowledge on this site. I wish you all the best going fwd.
3 Likes
I was a comparatively early adopter of rapamycin when this site had a much smaller membership. Dr. Blagosklonny was still alive and on Twitter at the time. The idea he had at the time was to take the highest dosage that did not have any serious adverse effects. No one has ever died from a really high dose, according to the literature I studied before I started. I started at 20 mg rather than titrating up, which is the recommended practice, and this caused me to have diarrhea the next day. I took 20 mg twice to make sure it was not a coincidence that it caused me to have diarrhea. After that, I titrated down to a dosage that didn’t give me diarrhea. 12 mg was the result. The crucial tremors went unnoticed since I was not taking rapamycin, believing it would heal anything. I just realized one day that I was missing them. One day while eating, I became aware that my hands were not shaking when I tried to put food in my mouth. I am not sure how long it took for the tremors to stop after taking rapamycin. However, it was about 5 or 6 weeks. I was taking the following doses, as I recall: 20, 20, 15, 12, 12, 12.
My hands are really solid, and the tremors have never come back.
6 Likes
adssx
#492
Just published on calcium: Calcium-mediated regulation of mitophagy: implications in neurodegenerative diseases 2025
Calcium signaling plays a pivotal role in diverse cellular processes through precise spatiotemporal regulation and interaction with effector proteins across distinct subcellular compartments. Mitochondria, in particular, act as central hubs for calcium buffering, orchestrating energy production, redox balance and apoptotic signaling, among others. While controlled mitochondrial calcium uptake supports ATP synthesis and metabolic regulation, excessive accumulation can trigger oxidative stress, mitochondrial membrane permeabilization, and cell death. Emerging findings underscore the intricate interplay between calcium homeostasis and mitophagy, a selective type of autophagy for mitochondria elimination. Although the literature is still emerging, this review delves into the bidirectional relationship between calcium signaling and mitophagy pathways, providing compelling mechanistic insights. Furthermore, we discuss how disruptions in calcium homeostasis impair mitophagy, contributing to mitochondrial dysfunction and the pathogenesis of common neurodegenerative diseases.
Intriguingly, evidence suggests that mutated α-synuclein can deregulate the function of Ca2+ channels and lead to increased cellular Ca2+ levels, excessive Ca2+ uptake by mitochondria and the collapse of their Δψm.
Supporting this notion, PD-causing mutations in PINK1 have been associated with mitochondrial Ca2+ overload in dopaminergic neurons.
ocalized increases in cytosolic Ca2+, particularly within microdomains between subcellular compartments, may regulate mitochondrial dynamics and motility, thereby predisposing mitochondria for mitophagy. These localized Ca2+ signals may also modulate key mitophagy components, facilitating subsequent steps in the process. On the other hand, increased Ca2+ influx into mitochondria and mitochondrial Ca2+ overload, often induced by damage, may serve as an initiating signal for mitophagy pathways. This suggests that local cytosolic Ca2+ fluctuations could act as a mechanism to regulate mitophagy in a manner aligned with broader cellular demands and conditions, while mitochondrial Ca2+ concentrations may function more specifically as indicators linking mitophagy to the status of individual organelles. This dual role underscores the complexity of the interplay between Ca2+ signaling and mitochondrial quality control mechanisms, emphasizing the need for further investigation to unravel these relationships.
Poke @John_Hemming: could menaquinone-7 benefit here if it improves calcium homeostasis?
Higher calcium intake (as well as dairies) is often associated with a higher PD risk. Same for vitamin D serum levels
2 Likes
If calcium is undermining Δψm maybe mk-7 would help in resisting this. I don’t know enough about the relationship between mk-7 and calcium to comment.
2 Likes
Thank you for that. I haven’t had time to get through the whole paper yet, but I am very focused on calcium channel signaling in NDD in general and PD in particular. Looking at the references in this paper they tag an older (2019) paper by E. Leandrou, and have not referenced a newer paper by her (2024), which has had huge impact on my thinking. I was trying to link PD pathologies in calcium handling to other neurodegenerative diseases and broaden it beyond the substantia nigra - and lo and behold I found exactly that in the Leandrou paper! My intuition was right! Here is that marvelous paper:
α-Synuclein oligomers potentiate neuroinflammatory NF-κB activity and induce Cav3.2 calcium signaling in astrocytes
“It is now realized that Parkinson’s disease (PD) pathology extends beyond the substantia nigra, affecting both central and peripheral nervous systems, and exhibits a variety of non-motor symptoms often preceding motor features. Neuroinflammation induced by activated microglia and astrocytes is thought to underlie these manifestations. α-Synuclein aggregation has been linked with sustained neuroinflammation in PD, aggravating neuronal degeneration; however, there is still a lack of critical information about the structural identity of the α-synuclein conformers that activate microglia and/or astrocytes and the molecular pathways involved.”
“The sustained NF-κB activity triggered the upregulation of astrocytic T-type Cav3.2 Ca2+ channels, altering the astrocytic secretome and promoting the secretion of IGFBPL1, an IGF-1 binding protein with anti-inflammatory and neuroprotective potential.”
adssx
#495
However:
The finding that astrocytic Cav1.2 channels are expressed in astrocytes in vivo is also important based on the use of isradipine, an L-type Ca2+ channel (LTCC) blocker currently approved as a drug for the treatment of high blood pressure, as a potential therapeutic approach for PD. However, despite the encouraging findings in pre-clinical models showing alleviation of the LTCC-mediated Ca2+ load in the dopaminergic neurons, isradipine failed to confer neuroprotection in a phase III clinical trial on early PD patients. Our study revealed that Cav1.2 channels are significantly decreased in the A53T Tg mouse model of synucleinopathy. In this paradigm, the use of selective LTCC agonists to restore Cav1.2 activity will allow us to investigate the involvement of this channel in astrocyte function in future studies.
I was told that isradipine would be tried again but in a subset of PD patients based on genetic stratification. Another approach that is also explored at the same time is intranasal isradipine as some researchers think that the trial failed due to low levels in the brain.
The authors conclude:
In this context, our work highlights Cav3.2 as a novel druggable molecular target to alleviate the damaging effects of microglial and astrocytic activation.
One company is exploring that:
Source
In general, all calcium channel blockers are associated with a lower risk of PD in association studies and MR, for instance:
Source
1 Like
No, not isradipine. With the focus on astrocytes, I was rather thinking if sodium benzoate, much more accessible:
Sodium Benzoate, a Metabolite of Cinnamon and a Food Additive, Upregulates Neuroprotective Parkinson Disease Protein DJ-1 in Astrocytes and Neurons
The other aspect of astrocyte pathology is connected with cholesterol production in the brain. I was trying to unravel why there’s been such iffy effects of statins on PD, and it occurred to me that perhaps by inhibiting cholesterol production in the astrocytes, the statins were deranging calcium handling in the astrocytes, and that’s one avenue I’m looking at at the moment; too early for any conclusions, but it looks promising.
cholesterol production is downstream of the main acetyl-CoA flows. An odd complication is that NF kappa B increases citrate flow from the mitochondria which results in higher cytosolic acetyl-CoA.
There’s a paper, but it’s Iranian, so I don’t know if it’s worth pursuing:
Current insights into pathogenesis of Parkinson’s disease: Approach to mevalonate pathway and protective role of statins
The effect of statins is to stop acetyl-CoA being used for cholesterol. That means there is more available for other things (like acetylating histones).
Yes, but why would there be differential effects on PD depending on the statin? It’s all the mevalonate pathway. But in the veterans study, simvastatin was protective, yet atorvastatin, lovastatin not. OK, that was a clinical finding, but worth asking what could possibly account for the difference from a biochemical pathway mechanism.
I haven’t really studied the statins, but AFAIK they have different effects in different tissues.
Certainly in the brain tissue, because there it’s the astrocytes that produce cholesterol, not the hepatic tissue for example. That’s why I feel calcium signaling in astrocytes is key, and perhaps somehow atorvastatin is disrupting it, while simvastatin is not. If one could figure out what the difference is, it would be a huge step forward in unraveling the PD pathology.