tongMD
#1473
When those side effects happen to those on high dose statins who are not vegans and non-statin-taking vegans (note high dose phytosterols from diet and lack of certain types of saturated fat intake, in general, can cut down LDL a lot, but there are potential issues with that approach in select individuals because phytosterols that do get absorbed are atherogenic) at the same time - I’m not sure how much certainty you can put to “confounding”.
Because there is very limited evidence linking low LDL levels with an increased risk of osteoporosis.
If you can show me any studies with a substantial cohort, RCT, and substantial time I will change my mind.
tongMD
#1475
The other option is to wait for more information while dynamically hedging your bets carefully. I size my bets accordingly to confidence levels with as much quality information from the strongest arguments of both sides to avoid overconfidence.
I’m also concerned about a whole other myriad of issues such as anxiogenic cholecystokinin tetrapeptide levels on depression and suicide (rarely mentioned) just as another example of how complex downstream cholesterol functions can get - among other issues. Again, the issue is a lot more complex than it may seem. Better to assess and evaluate risk carefully. I can’t say for you - but most people are bad at that - it’s like a vast majority will raise their hand in a room of 100 random people asked whether they are an “above average driver”. Almost half of people are clearly wrong and overconfident. The other half may be to some extent as well.
1 Like
A_User
#1476
There is some mendelian randomization results on this, and triglycerides seem to be causally associated, LDL only weak evidence:
There was consistent evidence that triglyceride (TG) is causally associated with DS (MR-IVW β for one-s.d. increase in TG = 0.0346, 95% CI 0.0114–0.0578), supported by MR-IVW and GSMR and multiple r2 clumping thresholds. We also observed relatively consistent associations of TG with DSH/suicide (MR-Egger OR = 2.514, CI 1.579–4.003). There was moderate evidence for positive associations of TG with MD and the number of episodes of low mood. For HDL-c, we observed moderate evidence for causal associations with DS and MD. LDL-c and TC did not show robust causal relationships with depression phenotypes, except for weak evidence that LDL-c is inversely related to DSH/suicide. We did not detect significant associations when depression phenotypes were treated as exposures.
Statins tend to lower triglycerides too. And apoB can measure it all.
tongMD
#1477
Just to keep it short - it’s not clear whether it is the statin or LDL lowering that is the cause as you may already know. Again, there are a lot of dimensions involved. Lipids aren’t in isolation - TG vs HDL vs LDL are dynamic to an extent. Not going to get into subfractions. Ultimately, the paper you put forth does not analyze nonlinear relationships.
To be clear, I realize the potential causative and potential confounders involved are complex, hence I adjusted mortality curves for all the potential known acquired low LDL reasons that confound mortality, treatments vs treatment naive, and compared it to known genetic factors to avoid biasing towards higher LDL targets for “healthy” with the assumption of the LDL hypothesis being true where apoB and LDL are a true causative independent factor.
I’m not going to go super in-depth here on the finer points, as this discussion can go on for a very long time and from previous experiences, it’s probably not particularly fruitful for everyone here (not just us) while the benefit from ARR theoretically is marginal assuming the average “healthy” individual and “perfect” lifestyle factors with say LDL 75 and favorable phenotype/no discordance. We’ll agree to disagree if you wish but I suggest one at least compare the theoretical ARR using the sort of validated 30 year Framingham data if one hasn’t yet. It’s a very simple exercise.
It’s your body and your choice. I’m merely describing what I do personally and how I got to it very generally speaking - I have a low risk tolerance. How one decides what to take or not take with their lipidologist is something they will have to decide on their own with their own risk tolerance levels.
I’ll just finally say the common theme I see with lipidologists is the more one reads, the less one thinks one knows. Maybe about half of what one reads ends up being meaningless, but we don’t know which half is the issue so to speak. One of my dad’s best pieces of advice to me before I started med school that keeps ringing true the deeper I get - and he has decades of experience managing clinical trials.
To reduce the risk of being dead wrong, I encourage one to read the newest edition of Therapeutic Lipidology or at least some other up to date text that is essentially equivalent and pick apart the potential errata or flaws in the text, if they haven’t already. Make sure to do one’s homework if one doesn’t. Most people don’t even want to do basic homework so take that into account.
3 Likes
Thank you for the schooling.
But, I think we have lost the thread of the conversation.
Are you saying that low LDL if it is in the recognized normal range by labs such as LabCorp and Quest Diagnostics is worse than higher LDL? And, should we ignore the fact that rapamycin raises LDL in many people?
If you want to prevent aging-related vascular diseases one of the first things to do is keep your lipids in the normal range. Most people with a good lipid profile will not be obese and are probably moderately active or exercise.
BTW, I did take probability and statistics in school. And, it is obvious to me many people do not understand the difference between comparative, relative, and absolute risk.
I am currently taking rapamycin, but the long-term risk factors have not been determined in humans. The long-term risk is not a major factor to me at 82, but, if I was younger I certainly would be keeping an eye on my cholesterol levels regardless of the fact that rapamycin proponents choose to ignore this. In another thread, we have been discussing the long-term negative effects of sleep supplements. The dementia problems don’t show up for decades unless you already have dementia.
Maybe this is the first warning:
[Rapamycin might be bad for the heart? - #2 by jakexb]
3 Likes
RapAdmin
#1479
For people really into this topic, there “might” be a free download of this textbook at this location: Libgen.rs sourced Textbook: Therapeutic Lipidology, Series: Contemporary Cardiology, Publisher: Humana, Year: 2020
I believe that Rapamycin produces some great benefits, but also has side-effects. Therefore additional treatments need to be added to reduce those side-effects in order to make Rapamycin more effective. This can be seen when Rapamycin is paired with either acarbose or metformin. Maybe a Rapamycin and statin pairing would also be of benefit.
Someone should mention this to Dr. Miller for a trial?
1 Like
tongMD
#1481
To be clear, I understand people have taken stats in school and I’m not trying to “school” anyone here - it’s rather to help folks understand how they can get involved in learning if they wish to and recognize a knowledge gap, if any. I suspect pretty much everyone here probably has a college education at least if not further - although one doesn’t have to go to college to learn this.
There’s also a difference between advanced stats where someone at a PhD level can easily pick apart incorrectly applied methodology (as well as most masters level of knowledge can probably do it with enough practice - again, one doesn’t have to go to college to learn this and I’m not intending to be exclusionary here) which happens all the time in medical research papers vs undergraduate stats & probability who probably won’t realize it most of the time, especially when applied to medical papers in certain labs without relevant context, assuming they aren’t reading just the title and abstract which seems to happen pretty often.
LCME for docs actually didn’t require stats/biostats back then for the curriculum - so it’s up to old docs who didn’t learn to keep up with CME. Same with docs with just regular biostats knowledge that did not go to a more advanced level. So this is not a doc vs not doc deal. It’s whether someone actually took the time to read through knowledge from textbook/papers with relevant knowledge/etc with understanding of relevant skills, regardless of whether they are a doc or not, and I suspect >9/10 it’s usually someone who does not have those relevant skills and background, even though I try not to assume too much to avoid underestimation, and I’m trying to be as inclusive as possible regarding any mutually interesting discussions because I enjoy the approach to crowdsourcing information and talking to folks on rapa.
I’m not saying low LDL is necessarily better or worse than higher LDL in isolation. It’s a complex situation, hence I mentioned “and favorable phenotype”. We should be concerned about the potential implications of increased LDL from rapa, but we don’t know for sure whether it is “bad” especially with the seemingly transient nature that varies by individual, as well as the apparent general phenotype.
1 Like
This is the takehome point for me. Thank you everyone who has been participating in this thread. I feel like its a masterclass in heart disease and possible strategies at risk minimization.
6 Likes
I assume Rapa’s mechanism to be an increase in mitochondrial efficiency which increases the Acetyl-CoA level in the cytosol (and as an effect of this the nucleus). That creates more substrate for the creation of cholesterol.
Hence one could expect more cholesterol of various forms.
A_User
#1484
To be honest, I don’t see the value in doing this. I understand if you’re a doctor or a researcher and you have to know as much as possible or fill in some possible knowledge gaps. If you think you’ve found a counter argument to anything said, just bring up that argument and we can discuss it. Asking those you’re debating with to start reading textbooks means you’ve probably run out of arguments. And that’s fine. I already said we don’t have to debate about 70 vs. 40 LDL. As I don’t have any arguments, except extrapolating from short term randomized trials (PCSK9i and statins), mostly.
1 Like
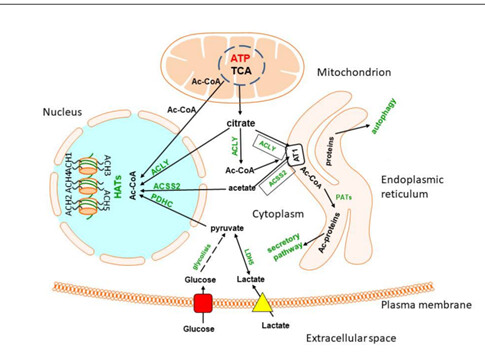
Potentially, but there are many , many ways for acetyl-CoA to be produced outside of the mitochondria. You have the ACSS, ACLY, PDH enzymes. You can have conversion of Acetate, lactate, NAA, glucose, pyruvate, citrate, etc.
Also those acetyl-CoA are not being used purely for cholesterol(as I am sure you are aware), they play a role in other things outside of lipid synthesis. It is also used for hundreds of acetylation reactions, including N-acetyl aspartate synthesis in neuronal mitochondria, acetylcholine synthesis in cholinergic neurons, as well as divergent acetylation of several proteins, peptides, histones and low-molecular-weight species in all cellular compartments.
Here is a good picture of some of the potential pathways for Acetyl-CoA( I study ACSS2 function in the brain).
Another thing to consider is where in the body acetyl-CoA levels increase. In the brain, it gets complex.
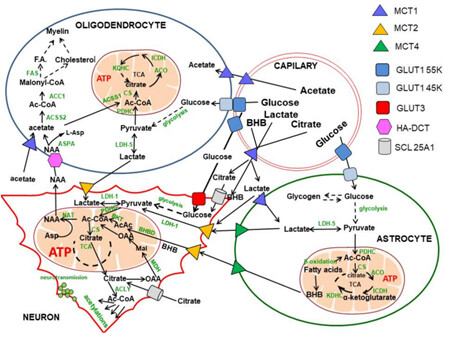
Point is, there are probably other factors involved in the increase of cholesterol we see with Rapamycin.
1 Like
ACLY, however, is fed by the non-canonical TCA cycle which is where mitochondrial health comes in. From the perspective of histone acetylation ACCS2 is interesting in that it is inhibited by growing levels of acetylation, hence if acetates increase acetylation will only go so far.
The reason I am particularly interested in Acetyl-CoA metabolism is that I believe this is at the core of two aspects of the key aging pathway which results in longer genes being expressed less over time. I wrote a blog post about this with links to the relevant research which is here:
In your studying about acetyl-CoA levels in the brain you may wish to look at my own experience. My biohacking experiments are designed to increase cellular acetyl-CoA (primarily to prevent the stalling of RNA Pol II and consequential aberrant RNA splicing). I am doing this on a cyclical manner where I intend to increase it in the morning starting roughly when I get up at 6am and stopping perhaps about 5pm, but obviously things go down more slowly than that. I do think it affects sleep in that if I stop the processes later in the day it is disruptive to my sleep.
The logic of this is by providing the substrate for HAT/KAT as part of the RNA Pol II complex there is less stalling. The macroscopic results are changes in biomarkers and the growth of new hair in various places as well as changes to some of the indicators of aging. The side effects that occur are the side effects that one would expect from providing more cytosolic Acetyl-CoA including a temporary increase in cholesterol although I have now driven down my LDL cholesterol levels although initially they were higher (although there are lots of problems with labs in measuring cholesterol). Also the availability of substrate for cytosolic ROS creation can result in inflammation although I have found it quite good at fighting infection and I think that in part is the cause of my WBC being low. Rapa pushes it down further, but I take Rapa quite infrequently.
1 Like
Another interesting thought I have just had based upon this conversation is that another thing that disrupts my sleep is menaquinone-7. (Vitamin K2 MK7). Now this molecule is interesting because it provides an additional electron transport for the mitochondria. Papers seem to indicate that it increases ATP production through OxPhos by about 5%, but I wonder whether that additional available energy has similar effects to the additional Acetyl-CoA. Whichever way additional energy tends to disrupt sleep. It may be that some of the additional Acetyl-CoA feeds back into the mitochondria to generate more ATP.
Correct on ACLY being fed non-canonical TCA cycle, but it also can take citrate produced through the TCA cycle and utilize that for Acetyl-CoA production, it can do the same with acetate. ACSS2 also recycles acetate that are released from HDAC reactions, allowing it to be used for Acetyl-CoA.
My focus has been on ACSS2 role in neuropsychiatric disorders and sleep. I recently conducted some experiments showing that inhibiting ACSS2 promotes resilience in social defeat stress model. My next step is to see how alterations in ACSS2 impact sleep homeostasis. I would imagine that changes in intracellular Acetyl-CoA levels will impact sleep homeostasis, since alot of our circadian clock genes are regulated through these epigenetic modifications, along with the fact you would get changes in sleep promoting gene transcription. Alterations in intracellular Acetyl-CoA pools have been shown in numerous neuropsychiatric disorders, as acetylation is a key regulator in immediate early gene function and neuronal plasticity and long term memory. I would imagine that too much acetyl-CoA will cause over transcription of genes, where some of those genes may be good, but others may not.
But if you want to talk about this more, we can have private message discussion or move this to another topic in the forum, because I think we are straying away from the main topic of focus.
1 Like
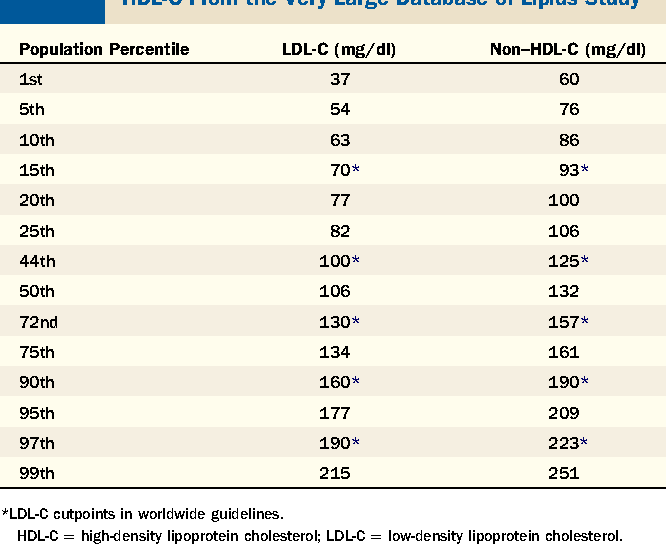
Non HDL is emerging as perhaps the most important lipid measurement. Here’s a very nice summary and suggests a non hdl of < 130.
The most important sentence is this, “ that said, it’s important to consider risk factors for cardiovascular disease on an individual basis rather than rely on the numbers.”
For the non clinician, it’s simpler to just stare at the numbers , but I suspect that the majority of rapamycin users are low risk and don’t need to jump to statins.
For those still concerned, bergamot is an excellent alternative
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4702027/
A_User
#1491
That is around the 50th percentile. I don’t think that is low enough as the average Joe or Jane gets heart disease. Under the 10th percentile would be 86 and under the fifth percentile below 76. There is plenty of benefit from going lower than the median.
I think levels around the normal levels of Acetyl-CoA tend not to have that much non-enzymatic acetylation.
1 Like