This paper has stuck in my mind for quite a while. So, with it gaining more attention lately, I contacted both the lead author and the senior author for clarification on a few points.
For context, the protocols in this paper were written over twenty years ago, with participants enrolling between October 2004 and November 2008. The lead author has also moved to a different institution in the interim. All that is to say, the original data is not easy to come by and both of them responded from memory. Here’s a summary of what I’ve garnered from the exchange:
The paper mentions that 6 of 12 participants exhibited a rebound of phospho-p70S6K. This data came from the sirolimus-alone study, which Supplementary Table 1 describes as having 40 total patients. How were these 12 subjects selected from the broader pool of 40?
Most Likely Explanation: The rebound analysis was performed in participants where viable measurements were taken at a sufficient number of time points.
The paper also mentions that the rebound was “driven by the concentration of sirolimus”. Does this mean that higher doses were more likely to cause a rebound?
Most Likely Explanation: Higher concentration = more rebound. No other characteristics (again, working from memory here) predicted a rebound.
I want to highlight that the lowest dose used in this wing of the study was 10 mg. The higher doses—the ones that predicted a rebound—were much higher. Here’s Table 4 from the paper:
| Dose cohort (mg) |
N |
Tmax, h (SD) |
Cmax, ng/mL (SD) |
AUC, ng x h/mL (SD) |
| 10 |
6 |
2.3 (0.01) |
9.67 (2.48) |
615 (130) |
| 20 |
7 |
2.3 (0.09) |
19.27 (5.69) |
1,084 (581) |
| 30 |
6 |
2.3 (0.16) |
43.47 (8.43) |
2,713 (1,346) |
| 60 |
8 |
2.2 (0.16) |
57.72 (22.6) |
3,142 (1,680) |
| 30, 4 hours apart (total 60 mg) |
5 |
2.2 (0.17) |
32.93 (10.7) |
2,054 (1,123) |
| 30, 24 hours apart (total 60 mg) |
3 |
2.3 (0.17) |
83.87 (18.8) |
3,677 (1,460) |
| 45, 24 hours apart (total 90 mg) |
2 |
2.1 (0.02) |
42.75 (0.08) |
3,356 (138) |
Here’s my takeaway: strongly inhibiting mTOR with high doses of rapamycin (e.g. 30 and 60 mg) can cause mTOR to rebound above baseline values. Lower doses (e.g. 10 mg) are unlikely to stimulate a rebound.
Why?
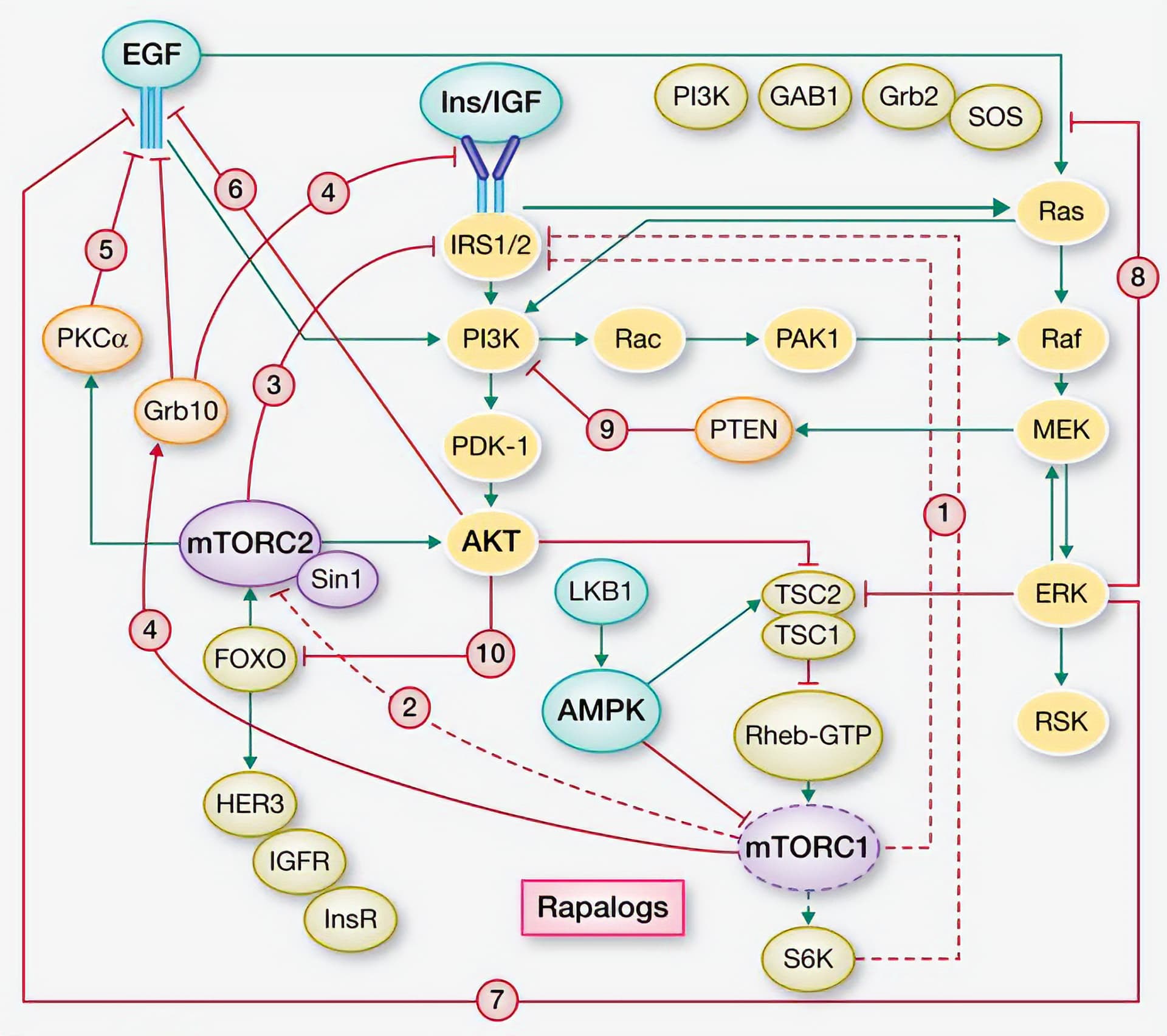
Under normal conditions, mTORC1 inhibits a variety of related signaling pathways, including: IRS1/2, mTORC2, and IGFR. Inhibiting mTORC1 with rapamycin releases this negative feedback (Rozengurt et al. 2014). These over-activated pathways can then stimulate mTORC1 through, for example, AKT and MEK/ERK. Take a look at this simplified diagram to get a sense of the reciprocal feedback loops at play here:
(Rozengurt et al. 2014)
The proteins highlighted in yellow are the ones that mTORC1 usually inhibits (and that are released by treatment with rapamycin). Please note that this figure is a useful simplification—there are far more interactions than displayed here. I can go into more detail in a separate post if anyone is interested.
It appears that high doses of rapamycin (e.g. 30 and 60 mg) repress mTORC1 activity low enough to activate a variety of compensatory mechanisms. Think of it as the cell’s attempt to maintain homeostasis by upregulating mTOR. I speculate that, as the concentration of rapamycin decreases, these over-activated compensatory mechanisms overshoot in the opposite direction, causing a surge in mTORC1 activity (i.e. the mTOR rebound).
In that same vein, lower doses of rapamycin (e.g. 10 mg) don’t repress mTORC1 activity low enough to set off the full spectrum of compensatory action. As @Krister_Kauppi mentioned, these rebound dynamics not unique to mTOR:
Summary: the rapamycin doses that caused an mTOR rebound in (Cohen et al. 2012) are much higher (up to 90 mg) than the doses most of us use. If you’re taking doses below 20 mg, you probably don’t need to worry about this.
If you’re still reading (and still concerned), then consider pairing rapamycin with metformin.
Metformin inhibits mTORC1 without releasing negative feedback loops and overstimulating AKT. It stimulates AMPK by inhibiting mitochondrial complex I. AMPK then phosphorylates IRS-1 (Insulin Receptor Substrate 1), whereas rapamycin suppresses IRS-1 phosphorylation. Metformin also inhibits MEK/ERK in the presence of growth factors, while rapamycin activates MEK/ERK by releasing feedback inhibition (Rozengurt et al. 2014). In male NcZ10 mice, combining rapamycin and metformin corrected for their independent downsides (Reifsnyder et al. 2022). Similar results were seen with 4 weeks of combination treatment in male Balb/c mice (4–6 weeks old) (Albawardi et al. 2023).